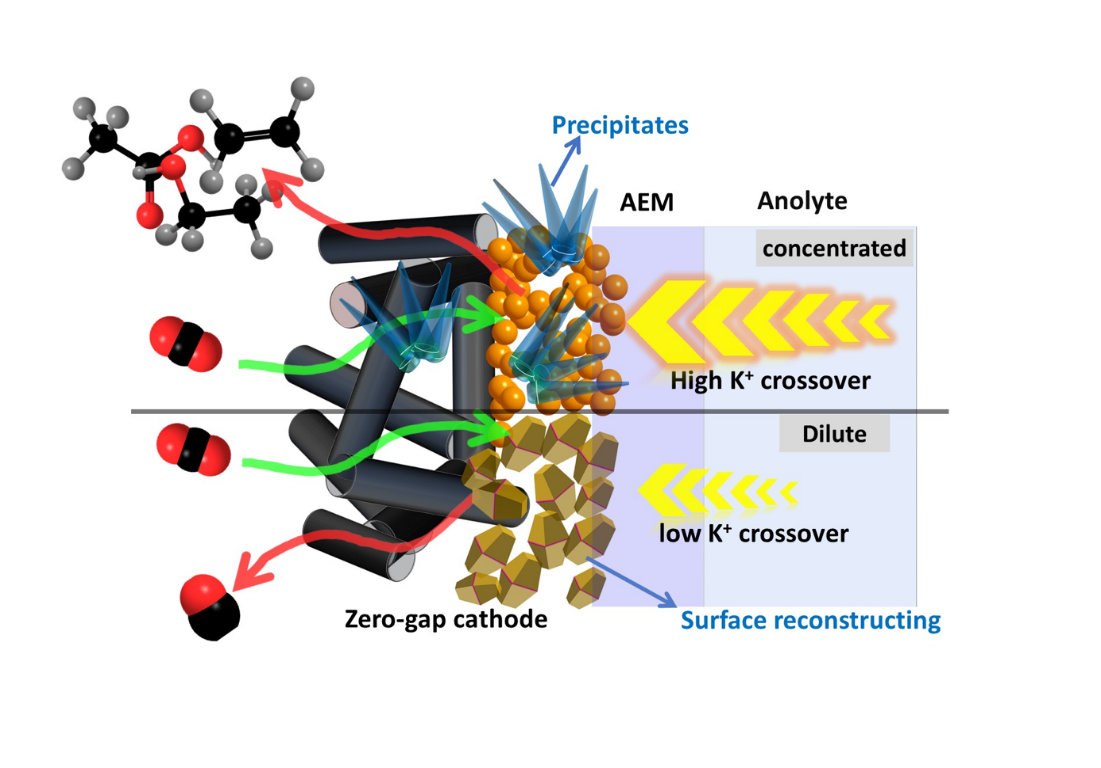
Anolyte concentration variation, constant voltage operation. For this study we employed a zero-gap GDE cell based on that reported previously by Endrődi et al.,15,16,21 here using commercial Cu nanoparticle (CuNP) catalysts spray-deposited onto commercial GDLs (see the Methods section for more details). As summarized in Fig. 1 and Supplementary Fig. S1, the cell stack comprised a cathode current collector with radial flow pattern, Cu-coated GDE, anion exchange membrane (PiperION), IrO2-coated-Ti anode, and anode current collector assembled in that sequence by compression. Humidified high-purity CO2 gas was fed to the cathode, while liquid anolyte from an external reservoir was pumped across the anode. No liquid catholyte was added. In CO2ER experiments using concentrated anolytes (KOH > 0.1 M) we observed formation of K-containing precipitates which contributed to performance degradation and cell failure, in agreement with previous reports. Since the flux of K+ through the membrane is likely correlated to its original concentration in the anolyte, we postulated that the use of more dilute anolytes might be one approach to mitigate the formation of precipitates at the cathode. Surprisingly, we found only a few previous studies which systematically varied the anolyte concentration for MEA CO2ER devices, mostly conducted using Ag catalysts, few exploring low concentrations < 0.1 M.16,30,31 We therefore conducted a series of experiments where the anolyte concentration [KOH] was systematically varied over a wide range, including concentrations < 0.1 M which normally impart prohibitive ohmic resistance when using catholyte-containing cells, but are uniquely enabled via the zero-gap configuration.
For each experiment, a pristine Cu-GDE was prepared and assembled into the cell. Each was tested using flows of KOH anolyte of different concentration, while gaseous products were measured by on-line gas chromatography. A two-electrode configuration was used, since in the absence of catholyte it is difficult to incorporate a reference electrode for controlling or measuring the cathode potential. Thus, voltages reported here correspond to bias between the anode and cathode current collectors. In the first experiments, a fixed bias was applied to the cell (vide infra for constant-current experiments). Besides the varied anolyte concentration, all other system parameters were kept constant. See Supplementary Note 1&2 for description of the experimental sequence. Figure 2a depicts the measured faradaic efficiency (FE) toward major products as a function of [anolyte], as determined by repeated gas chromatograph (GC) injections. At higher values (≥ 0.1 M), typical behavior for Cu is observed – C2H4 is the dominant product along with the H2 evolution side-reaction, and CO formation is minimal. A variety of other products are also detected but omitted here for clarity (see Fig. S2a for complete characterization). In contrast, using [anolyte] < 0.1 M resulted in a drastic change in selectivity – CO became the dominant CO2ER product while C2H4 was suppressed. At 0.05 M KOH an optimum CO selectivity was observed, with FE over 75% while H2 was under 5%. Interestingly, the selectivity and formation rate of C2H4 decreased progressively with decreased anolyte concentrations. Even using pure water without any (intentional) alkali metal cations resulted in selective formation of CO, albeit accompanied by significant H2.
This striking trend shows that anolyte concentration can strongly impact cathode selectivity and activity for MEA CO2ER, and we show here for the first time that this is especially significant when using Cu catalysts normally capable of forming many different products. As stated above, the only parameter varied was the concentration of KOH used as anolyte, which we hypothesized could influence the flux of K+ across the membrane. To assess this, inspired by past studies where aliquots of liquid water were used to rinse the GDE during operation, we performed in situ rinsing of the GDE to collect and quantify K+ during operation. For each [KOH] experiment in Fig. 2a, after continuous CO2ER operation for 60 min, an aliquot of pure water was injected into the gas stream, forced through the GDE flow channel and out of the cell, and collected a downstream trap for subsequent [K+] quantification by ICP-OES (as described in detail in S1 note #3). While not a quantitative measure, this approach allows us to assess the relative content of soluble K+ accessible at the gas diffusion side of the electrode following extended operation under the different conditions. The moles of collected K+ per electrode surface area are shown in Fig. 2a (right y-axis), revealing a clear trend which coincides with the inflection of the selectivity – for [KOH] < 0.1 M the detected K+ is low and varies only gradually with [KOH], whereas there is a steep increase across higher concentrations reaching significant magnitudes of K+. This result gives supporting evidence that the amount of K+ reaching the cathode is likely a determining factor in the observed selectivity trends.
It must be acknowledged that although the anolytes began as pure KOH solutions, during cell operation under CO2ER conditions they transform significantly. The homogeneous reactions of CO2 with OH− (electrochemically-generated or from the anolyte) produce HCO3− and CO32−, which readily move from the cathode through the AEM and gradually transform the anolytes into KHCO3 solutions when the total electrolyte volume is fixed and recirculated through the cell.18 Evidence of this is seen in anolyte pH measurements vs time (Fig. S3a), where the pH values shift toward values expected for carbonate/bicarbonate buffer. This behavior supports that carbonate is the main current-carrying ion across the membrane, and the neutralization rate depends on the current density (2 e− lead to neutralization of one KOH) and the initial amount of KOH. For more detailed discussion of the combination of processes producing this pH change we refer the reader to some key recent studies.18,32 Despite this, most of the results presented herein employed KOH solutions as the starting anolyte, a decision based on the predominant use of alkaline anolytes in the recent CO2ER literature achieving high device activity. Later, we performed analogous experiments using KHCO3 anolyte and found similar trends (vide infra).
For measurements of Fig. 2a conducted at constant cell bias of 3.2 V, the resulting total current densities are shown in Fig. S3b. In each case, the currents show some initial transient behavior likely corresponding with the aforementioned anolyte pH transformation, but after ca. 20 min. they each remain stable over the course of the testing period. Once the pH and current densities stabilize, the devices using [KOH] ≤ 0.1 M exhibited stable operation and produced consistent product selectivity over repeated GC injections. At higher concentrations, the current densities were less stable, likely due to precipitation-induced deactivation. Notably, the magnitudes of the current density scale with the anolyte concentration. This could be influenced by differences in ohmic resistance (across the electrolyte and membrane) or by differences in electrode activity resulting from the varied electrolyte concentrations. Since no reference electrode was present, it is challenging to deconvolute the precise origin of the variation. To help understand the cell behaviors, electrochemical impedance spectroscopy was conducted on the cells during operation, and the Nyquist plots are provided in Fig. S3c. The high frequency intercepts correspond to ohmic resistance in the cell, which were low and rather consistent (0.18–0.22 Ω) despite the wide variation in anolyte ionic strength, suggesting the zero-gap configuration minimizes the sensitivity to electrolyte conductivity.11 In contrast, the charge transfer resistances (estimated by comparing the diameter of the semicircle features) do vary, generally decreasing with increased ionic strength. This parameter is sensitive to the kinetics of the electrode processes, suggesting that processes on the anode and/or cathode are influenced by the presence of cations. In MEA devices, precise deconvolution of the contributions to the 2-electrode cell response is challenging and can be can be aided by numerical modeling, although to our knowledge previous models for MEA CO2ER have not included the effects of anolyte neutralization in alkaline MEA configurations since they assumed an infinite and rapid feed of alkaline anolyte.11
Constant current operation. It is well known that Cu catalysts typically show potential-dependent selectivity,4,9 and thus activity comparisons would ideally be conducted under fixed cathode potential. But the zero-gap configuration makes the use of a reference electrode challenging.33 Comparisons on the basis of rate-controlled experiments are equally important, to normalize by the kinetics of cell operation. We therefore conducted controlled-current experiments using cells with 0.05 M and 1.0 M KOH anolytes (conditions favoring CO and C2H4, respectively). Single Cu-GDE cathodes were used for each condition, stepped progressively from low to high current, at the end of each step injecting a H2O aliquot for K+ crossover determination. As shown in Fig. S4, when varying the current density across the range 38–140 mA cm− 2, CO persisted as dominant product in the 0.05 M KOH cell. Measurements of K+ from cathode rinsing maintained low values as the current increased, while CO remained the dominant product. As current increased the C2H4 formation rate gradually increased. In the electrolyser operated with 1.0 M KOH, the measured K+ was much higher, and C2H4 and H2 were the major products. The CO yield remained very low across all currents tested, even at the lowest currents which correspond to low bias voltage, which is an important observation since it shows that the observed CO/C2H4 selectivity switch is not likely simply due to differences in applied potential at the cathode. Thus, across a range of currents and voltages, the concentration of the anolyte, and resulting flux of K+ to the cathode, appear to be the key determining parameters for the selectivity switch.
Electrolyte switching to evaluate reversibility. At this point, there arises the question of whether the observed effects of K+ on the cathode reactivity are reversible, i.e., whether the selectivity trends will switch when the electrolyte is exchanged. If water-solvated cations which crossed to the cathode are responsible for the selectivity trends, then the activity may be switchable by changing the electrolyte. To examine this, we conducted an experiment in which the cell was first operated with 1.0 M KOH anolyte for 10 minutes followed by exchanging anolyte with pure H2O. Figure 2b shows the resulting product selectivity over time, where the starting behavior correlated with the 1.0 M case of Fig. 2a (primarily C2H4 and H2 with minimal CO), then gradually but significantly transformed toward CO production following the switch to H2O anolyte. This result suggests the cation effect is indeed reversible, and that K+ can diffuse away from the cathode when the concentration gradient is reversed. This suggests a dynamic equilibrium between anolyte, membrane, and cathode impacts the distribution of cations across the cell, and that cations are not continuously driven to the cathode by the electric field or as primary charge-carrying species (which is also supported by the drastically different K+ amounts detected when comparing experiments at fixed current densities, Fig. S4). Note that although the KOH solution was exchanged with pure H2O in the anolyte reservoir, residual K+ persists in the system which likely contributes to the long-term behavior of the device, producing mainly CO with keeping both H2 and C2H4 below 10%.
Origin of cation transport dependence on concentration. The above control experiments thus established that the observed switch in selectivity between CO and C2H4 (Fig. 2a) is not simply arising from differences in applied potential or current density, but rather seems mainly to be dictated by the degree of K+ crossing the membrane to interact with the MEA cathode. We consider this to be an important observation for the research community working on catholyte-free devices for electrocatalytic reactions, and a particularly exigent issue for those working on Cu-based devices for electrochemical conversion of CO2 and CO. As discussed in the introduction, alkali cation effects are well known to influence copper’s CO2ER selectivity, but to date little attention has been paid to how this translates to MEA-based devices. While many past studies have observed evidence of cation crossover through AEMs during operation in the form of solid precipitates, little attention has been paid to exactly how and why the membranes allow such significant flux of co-ions which are expected to be excluded in ion exchange membranes. Species transport across membranes is a rather complicated matter, affected by many parameters including concentration gradients, electrochemical potentials, current densities, water uptake, membrane morphology, and so on,11,34−36 and a detailed understanding is outside the scope of the present work. But at this juncture we wish to emphasize one basic principle governing the permselectivity of ion exchange membranes: the Donnan exclusion principle.37–39
As a general explanation, Donnan exclusion relates how the capacity of an ion exchange membrane to exclude co-ions (ions of same charge polarity as membrane ionomer charge, e.g. cations to an AEM) from an electrolyte solution depends on the relative density of charges between the two phases – for the membrane, the activity of fixed-charge groups (i.e. ion exchange capacity); for the electrolyte, the activity of dissolved salt (i.e. ionic strength). When the electrolyte concentration is comparable to the membrane’s fixed charge density, minimal Donnan exclusion exists, resulting in poor permselectivity based on charge type.38 In practice the situation is surely more complex, but this principle can immediately guide us to the understanding that the transport of cations through an AEM will be a function of the electrolyte concentration, and this distinctly correlates with our observations in this study. The PiperION AEM used here has a fixed charge density of approx. 2 M,40,41 and we observed that K+ readily reaches the cathode when using anolyte concentrations in this regime. Only for concentrations ≤ 0.1 M is K+ strongly excluded. Important to note is that ion exclusion is never perfect, and this is crucial to consider when considering reactions like CO2ER which are so sensitive to the presence of alkali cations.29 Indeed, based on the trend we observed as the anolyte concentration approached zero in Fig. 2a – CO yields dropped as H2 rose – it appears that some amount of K+ reaching the cathode seems necessary for optimal CO selectivity.42 Due to the dynamic nature of ion movement and the close integration of membrane and GDE here, we cannot precisely quantify the cations at the electrocatalytic interface, but this is a goal for future study.
To closer investigate the combined effects of concentration and electrochemistry on the magnitude of K+ crossover, we conducted a series of tests where the cathode was rinsed repeatedly to sample K+ over an extended period. The cation quantification is shown in Fig. S5. Note that since the act of repeated rinsing itself changes the amount of cations at the cathode, this data is used for qualitative observation of trends only. First, comparing cells operating under CO2ER conditions with concentrated and dilute anolytes, we see that even in the first minutes, the 1 M KOH leads to K+ levels more than two orders of magnitude greater than the 0.05 M case. During extended operation, the concentrated anolyte gives consistently high values but with irregular behavior, likely due to the formation of significant solid precipitates throughout the GDE which are incompletely collected by each rinse. The dilute anolyte also showed some increase in K+ crossover with time, but never reached high values. For comparison, a cell with 1 M KOH was assembled and allowed to simply sit at open circuit, repeating the cathode rinsing procedure. In this case, the K+ crossover remained low, far below that of the same configuration under CO2ER conditions, and interestingly, comparable to the case of 0.05 M cell under operation. This suggests that electrochemical conditions also play an important role in activating K+ crossover. This could arise from several factors but in the simplest sense one invokes the Donnan principle again – the interfacial Donnan potential (at the membrane-solution interface) acts to exclude co-ions but it is modulated by the external bias. Therefore, electrolyte concentration and electrochemical potentials must be considered to fully understand permselectivity. A complete investigation of these effects is beyond the scope of this report, but these points deserve deeper attention in the electrosynthesis community, and we refer interested readers to some key literature.35–39, 43,44
Extended operation under CO-producing conditions. The configuration yielding optimal CO selectivity (0.05 M KOH) was tested under constant bias for an extended period of continuous operation (Fig. 2c). After an initial equilibration period, the CO FE reaches 80% and remains stable over the course of five hours, while the yield of C2H4, H2, and all other gaseous products remains low, accounting for less than 10% FE. Liquid products were quantified after the experiment and found to be negligible. The stable product selectivity and current density over extended operation suggests that this alkali cation-deficient configuration can operate under equilibrium where the cathode microenvironment favors CO production, without flooding, precipitation, or loss of conductivity.
Structure and speciation of catalysts. Selectivity trends for Cu-based catalysts are often correlated to surface speciation (e.g., Cu oxidation states) and/or morphology, as well as how these factors may evolve during CO2ER.4 Although in our experiments above every electrode was prepared identically, it is possible that the use of different anolytes affects the resulting active form of the Cu catalysts. Thus, we conducted in situ x-ray absorption spectroscopy (XAS) and glovebox assisted x-ray photoelectron spectroscopy (quasi in situ XPS), in addition to pre- and post-electrolysis SEM and GI-XRD to assess both aspects as function of employed anolyte.
Figure 3 shows SEM images of the as-prepared Cu catalyst layer in comparison with samples after CO2ER testing using different anolytes. Supplementary Figs. S6 and S7 provide additional cross-section and element mapping images. We observed that Cu tested with 1.0 M KOH anolyte showed microstructure (grain size and porosity) which resembled the as-prepared state, without significant morphological differences (Fig. 3b). More significant changes were observed for the cathodes operated with 0.05 M KOH anolyte (Fig. 3c), which exhibited a significant increase in average grain size. Additionally, the cathodes operated with concentrated anolytes (≥ 0.1 M) exhibited areas of K-containing wire-like precipitates (Fig. 3d), as indicated by their elemental mapping analysis (Figs. S6, S7).
XRD analysis of the samples indicated that their bulk is mainly composed of Cu and Cu2O (Fig. 3e). Although susceptible to oxidation in air, we can see here that samples tested using different anolytes seem to show different relative contents of oxide, which we investigate closer by spectroscopy below. These analyses showed that the anolyte significantly impacts the structure and morphology of the zero-gap cathode, despite catholyte-free configuration and AEM separating the cathode from the anolyte.
To examine the catalyst under true operating conditions, we conducted next in situ XAS to track the catalyst and its chemical composition during CO2ER. Cu K-edge XAS resulted in spectra which closely resemble Cu metal standard, regardless the anolyte concentration used, suggesting the complete reduction of the bulk material (within the limit of detection of this bulk-sensitive method; see Supplementary Note 3 for discussions of bulk vs surface sensitivity). Notable here was that the zero-gap configuration allowed design of an in situ cell which had nearly the same geometry as the laboratory cell used for CO2ER studies, which we validated under CO2ER operating conditions to give comparable current density and product selectivity (Fig. S9), meaning such a cell can be used for study under true operando conditions.
While hard X-ray XAS allowed operando study, the signals are bulk-dominated and therefore inadequate for gaining precise information about the catalytic interface. A complimentary surface-sensitive technique, so-called “quasi in situ” XPS, was used for better evaluating the surface speciation as function of employed anolyte. Here, cells are subjected to CO2ER inside an inert O2-free glovebox, followed by disassembly and transfer of the Cu-GDE to the XPS analysis chamber under vacuum without air exposure using a gastight transfer arm. The results are summarized in Fig. 4 and Fig. S8. The Cu Auger region was used for deconvoluting and quantifying copper speciation (Fig. 4a).45 The surfaces of the as-prepared Cu cathodes (exposed to air) are exclusively composed of CuO species (Fig. 4b), as expected for Cu metal with native oxide layer. For the Cu cathodes operated at 3.2 V and flowing 1.0 M KOH anolyte (C2+ products-selective), the oxide features disappeared entirely and the surface appeared fully reduced, while ones tested using either pure H2O or dilute anolyte (CO-selective cathodes) exhibited a mixture of Cu2O (35–45 at. %) and metallic Cu (65 − 55 at. %) surface species. As a general trend, we found that the surface metallic Cu species increases, while oxidized species decrease, along with increasing anolyte concentration (Fig. 4c). This highlights a possible role of the crossed-over cations on the cathode surface speciation. Moreover, the Cu cathodes operated with dilute anolyte exhibited much higher Cu2O surface species (even after days of air exposure) compared to operated cathodes with 1.0 M KOH.
KHCO 3 anolyte operation. As discussed above, recirculated KOH anolytes eventually become neutralized by reaction with CO2 to form (bi)carbonate solutions. For comparison, we performed additional experiments directly using KHCO3 solutions as starting anolyte. A similar CO2ER behavior as a function of anolyte concentration was observed for the bicarbonate system (Fig. S12a) where dilute anolytes led to CO production, while ethylene was favored for the concentrated anolytes. A noteworthy observation is that the threshold for selectivity switching occurred at comparatively higher anolyte concentration than for the KOH case. Extended operation of a cell using 0.1 M KHCO3 anolyte showed stable CO generation and only low C2H4 and H2 formation over a several-hour test (Fig. S12b). The similarities observed between using KOH and KHCO3 anolytes of varied concentration provides further evidence that the cation concentration is a main determining factor influencing selectivity trends.
Origins and implications of cation crossover. Previous studies of CO2ER MEAs commonly observe K+-containing precipitates under certain operating conditions, and models have been developed to predict threshold current densities at which the carbonate generation rate brings the K2CO3 concentration beyond the solubility limit.11 But the specific impacts of unintended alkali metal cation crossover on the CO2ER reactions, and the factors affecting the magnitude of the crossover, have received less attention in studies of Cu-based zero-gap cathodes. Meanwhile, the significant impact of cations on the CO2ER mechanism on Cu has been extensively studied in H-cell configuration experiments and studied by computational methods (e.g. DFT).24–26, 46 Thus, to date there has remained a disconnect between the sub-fields of mechanistic investigation and practical cell development. Our results highlight the importance of quantifying (and ideally controlling) the movement of cations from anolyte to MEA cathodes. The information gained from various techniques in this study indicate that the observed selectivity trends are not simply explainable by morphological/structural effects or by differences in electrochemical conditions. Our observations reveal major influences of the unintended cation crossover from anolyte to the zero-gap cathode on the CO2ER performance, even in catholyte-free AEM based electrolysers.
While the principles governing membrane permselectivity are generally established,34,36,38,39 it appears that many (or most) demonstrations of CO2ER using AEMs with liquid electrolytes are operating under conditions where co-ion exclusion is not expected to be significant – i.e., when using electrolytes of high ionic strength in comparison to the density of fixed charges in the membrane. One must not simply assume perfect ion selectivity occurs when using ion-exchange membranes, and that careful consideration of the membrane’s influence on the cathode reaction environment is critically important among the multitude of factors affecting CO2ER selectivity.34
Some reports indeed mention that overcoming Donnan exclusion can lead to co-ion crossover,11,30 but without providing detail of what this means in practice, or how it might guide electrolyte choices. Based on our results and general calculations of Donnan exclusion,38,39 significant exclusion occurs only when using electrolytes which are substantially more dilute (≤ 0.05 M) than those typically employed by researchers in this field. Importantly, this was enabled using a zero-gap MEA device where the decreased electrolyte ionic strengths did not contribute significant ohmic losses, and thus this cell configuration opens possible new avenues of research in controlling ion transport, once a better understanding is developed. Furthermore, while this configuration opens the attractive possibility of operating CO2ER devices with simply a pure water feed (via liquid or humidified gas), our results (and recent results of others)16,29 suggest CO2ER activity is hindered in the absence of alkali cations.
Thus, a complete understanding of AEM-based cells for CO2ER requires detailed study not only of anions and carbon balance, but also cations, since the membranes seemingly fail to exclude cations under most conditions reported to date. Recently several strategies have emerged based on the use of bipolar membranes or tailored ionomers to manage and manipulate ions and local pH,28,31,47 aimed at optimizing the local environment and minimizing CO2 losses. Even in those configurations, we recommend that researchers pay close attention to the possibility of unintended ion transport. On a related note, Donnan exclusion has recently been invoked in some studies where the catalyst microenvironment is tailored using different ionomer coatings to modify the CO2ER selectivity of Cu in an H-cell configuration.48,49
Once alkali cations reach the Cu cathode, they clearly influence the reaction pathways. Despite numerous previous studies of cation effects on aqueous CO2ER, to our knowledge none of them identified the selectivity trend (switching between CO and C2+ products) we observed here, nor did they identify an influence of cation concentration on Cu surface speciation. The uniqueness of our observations likely stems from the use of a zero-gap cathode, as compared to the aqueous H-cell configurations used by the previous studies of cation effects. In those studies, using bulk catholytes in direct contact with the catalyst, it may be that in each case the local environment contained cation concentrations already exceeding the threshold necessary for triggering C2+ formation and/or CO suppression. Few past studies of Cu catalysts using liquid catholyte have systematically examined the effects of a wide range of varied ionic strength, especially including ones as low as we explored here, likely due to the significant ohmic resistances which develop across dilute electrolytes which would require exceedingly large cell voltages in H-cells (or catholyte GDE cells). Nevertheless, even at low bulk concentrations in liquid solutions, electrolyte cations can accumulate at the interface with a polarized cathode. In this regard, the zero-gap configuration provides a unique opportunity to operate cells with very dilute electrolytes and to potentially limit the cations reaching the cathode interface.
While a detailed mechanistic understanding of cation effects is outside the scope of this work, our observations that selectivity switches between CO and C2H4 as a function of K+ reaching the cathode can be interpreted against existing hypotheses about CO2ER cation effects. Overall, three main hypotheses have been debated in the literature to explain cation effects: (i) stabilizing the reaction key intermediates via electrostatic interactions, (ii) regulating the local CO2 concentration by buffering the interfacial pH and (iii) tuning the local electric field.24–26 An experimental-theoretical study by Resasco et al. highlighted the essential role of cations in facilitating the C-C coupling step, and hence enhancing the selectivity towards C2+ products.50 They reported higher C2+ production rates in the presence of heavier alkali cations attributing to their higher concentration (due to more compact hydrated radius) compared to the lighter ones in the outer Helmholtz plane. In a related direction, Huang et al. reported that the enrichment of cation concentration near the electrochemically active sites facilitates CO2 activation and C-C coupling in acidic medium.28 They also observed H2 selectivity to scale inversely with [K+], similar to our observations across a range of low anolyte concentrations. Conversely, at higher concentrations we see H2 begin to climb with [K+]. In studies on Au in alkaline media, Goyal et al. observed an increase of hydrogen evolution with the increase of the near-surface cation concentration, deducing that high cation coverage enhances the rate of the sluggish Volmer step.51 Hence our observed effects of K+ on CO vs C2 selectivity, as well as H2 evolution, generally follow trends demonstrated in aqueous H-cell studies, although in the absence of three-electrode measurements and extensive controlled current experiments, it is challenging to delve quantitatively into the selectivity trends to propose detailed mechanisms at this time.
We note that in most previous studies investigating alkali cation effects on CO2ER, the common approach was to vary the identity of the cation (i.e., Na+, K+, Rb+, Cs+), whereas fewer studies explored varying the cation concentration. Doing so is understandably challenging since this would typically require also varying the anion concentration, which would result in pronounced differences in pH buffering effects which significantly affect CO2ER as well.52 Furthermore, due to electrolyte conductivity limitations, low alkali cation concentrations can only be examined under special conditions. A few very recent reports provide insight into effects of concentration and coverage of alkali metal cations. Monteiro et al. recently studied the effects of cation concentration in acidic electrolytes, wherein conditions of low (or no) alkali cations can be attained.29 Their results suggest that CO2ER is indeed sensitive to the concentration of cations, even in very low concentrations, and they reveal the key observation that no CO2 conversion is detected in the absence of metal cations in solution. Ovalle et al. showed experimentally that increased coverage of alkali metal cations correlated with an increased rate of CO2ER to CO on Au.53 Additionally, Huang et al.28 and Gu et al.27 recently separately showed that in acidic solutions alkali cations help suppress H2 evolution and promote CO2ER in GDE cells with catholyte.
In contrast to aqueous cell studies, reports identifying cation effects on MEA reactivity are more rare, and mainly use Ag-based catalysts for CO2 conversion to CO. Romiluyi et al. studied Ag MEAs with varied CsHCO3 anolyte concentration in the range 0.1-1.0 M, acknowledging that Donnan exclusion can be overcome with increasing concentration, and observing some anolyte-dependent sensitivity in the selectivity and partial current densities toward CO and H2 which they attributed to the presence of hydrated Cs+.30 Endrődi identified the importance of alkali cations on the activity of Ag toward CO production, and developed an approach to intentionally dose cation solutions into the GDE in order to activate and regenerate its activity.16 Their results, combined with our current observations and the above-mentioned studies conducted in acidic media, all point to the importance of alkali cations in the catalyst environment for activating CO2ER pathways.
Finally, we consider possible explanations for the Cu surface speciation trends we observed by XPS as a function of anolyte concentration. Although bulk copper oxides are readily reduced during CO2ER under all anolyte conditions (as evidenced by our operando XAS analysis), the surface sensitive XPS showed significant differences in Cu oxidation states after the reaction. If this was simply a result of the Cu surface being exposed to different pH environments at open circuit after testing, one would expect the most alkaline electrolytes to yield the most oxidized Cu (based on Pourbaix trends). In fact, we observe the opposite – as shown in Fig. 4c, concentrated anolyte resulted in more reduced Cu, while more dilute anolytes yielded more oxidized Cu. One possibility is that local cations are influencing the interfacial pH at the catalyst, wherein hydrolysis of hydrated cations provides a buffering effect on local pH.51,54 In that case, an environment with higher K+ concentration would produce a stronger buffering effect, keeping the local pH lower and thereby preserving metallic Cu, in comparison to dilute solutions with weaker buffer capacity which could allow pH rise and Cu oxidation. This could also influence the observed variation in surface morphology. Furthermore, it has been shown that alkali metal cations can affect morphology evolution due to cathodic corrosion via preferential interactions with different facets.55 It is also possible that the differences in operating current or cathode potential contributed to our observed speciation and morphology differences, and future experiments integrating a reference electrode can help deconvolute these factors.
This study identifies that under certain conditions, even pure Cu catalysts can exhibit selective CO production. This could present an interesting avenue to develop Cu-based CO2-to-CO electrolysers as alternative to more complex bimetallic compositions56,57 or rare metals typically regarded as the best CO-selective catalysts (i.e. Ag, Au), although here Cu is not a particularly good catalyst in terms of activity. Nonetheless, as pointed out by previous studies,19 this device configuration using AEM is prone to low CO2 utilization due to (bi)carbonate formation and crossover to the anode chamber. Further innovations in reactor design towards addressing this carbonate problem are necessary.28,58−60 In general, careful studies should be devoted to understanding the role of the membrane in CO2ER,34 and to investigating the influence of its physical properties (thickness and polymer backbone) on the ability of cation crossover. Additionally, operando mapping the cation movements and speciation as function of membrane type, time, and applied bias using operando tomography/radiography will be valuable for further development of AEM-based electrolysers. Conducting microkinetic and DFT studies to evaluate the impacts of cation surface coverage on the local environments near the catalyst surface and its influences on stabilizing key intermediates will help in understanding the role of the cations on CO2ER.
{kind=link}