Itgβ8 is mainly expressed in regulatory T cells in tumors
In vivo, the activation of TGF-β1 is largely dependent on integrins, including the αvβ8 integrin, whose expression is regulated by that of the β8 subunit (Itgβ8) 11 15. In order to understand the mechanisms leading to the activation of the latent complex in the tumor, we first analyzed Itgβ8 cellular expression in the TME.
To monitore Itgβ8 by flow cytometry, we took an unbiased approach by generating an Itgb8-td-Tomato reporter mice, in which we previously validated that td-tomato positive cells expressed Itgb8 protein in different cell types, including T lymphocytes 16. Flow cytometry analysis of tumors (melanoma and breast cancer) revealed that among host cells composing the TME, Itgβ8pos cells were mainly (85–95%) CD45pos hematopoietic cells (Fig. 1A-B). T lymphocytes (CD3pos), and particularly the CD4pos Foxp3pos (Treg) subset, composed the main portion of hematopoietic cells expressing Itgβ8, with approximately 80% of Itgβ8pos CD45pos cells being CD4pos Foxp3pos irrelevant of the tumor type (Fig. 1C-F). Moreover, within the Treg compartment, we found that about 40–45% of cells expressed Itgβ8 (Fig. 1G-H) and only Itgβ8pos Tregs were endowed with the capacity to efficiently activate TGF-β1 (Figure 1I) whereas both Itgβ8pos Treg and Itgβ8negTreg populations expressed similar levels of this cytokine (Figure 1J). Thus, this first set of data reveals that Tregs constitute a large part of the Itgβ8-expressing host cells within the TME.
Itgβ8 expression in Tregs impairs anti-tumor response and promotes tumor-growth
Next, in order to assess whether Itgβ8 expression by Tregs confers them abilities to control the anti-tumor immune responses by providing a bioactive source of TGF-b, we first selectively ablated Itgb8 in Tregs, using Foxp3-Cre Itgb8fl/fl mice (Foxp3ΔItgβ8). Importantly, in Foxp3ΔItgβ8 mice Tregs retain their numbers, localization, as well as their suppressive functions, including the ability to produce TGF-β1. Moreover, no autoimmunity signs, neither uncontrolled effector T cell activation have been observed in Foxp3ΔItgβ8 animals 17, 18.
Strikingly, in contrast to their littermate controls (Foxp3Ctrl), Foxp3ΔItgβ8 mice showed a profound impairment of tumor growth irrelevant of the tumor type (Fig. 2A-F). Notably, we observed that 25–50% of the Foxp3ΔItgβ8 animals exhibited a complete control of the tumor progression depending of the tumor type (Figure G-H). Thus, Itgβ8 expression in Tregs promoted tumor growth, implying that the Itgβ8pos Treg population could affect the anti-tumor function of the effector T cells.
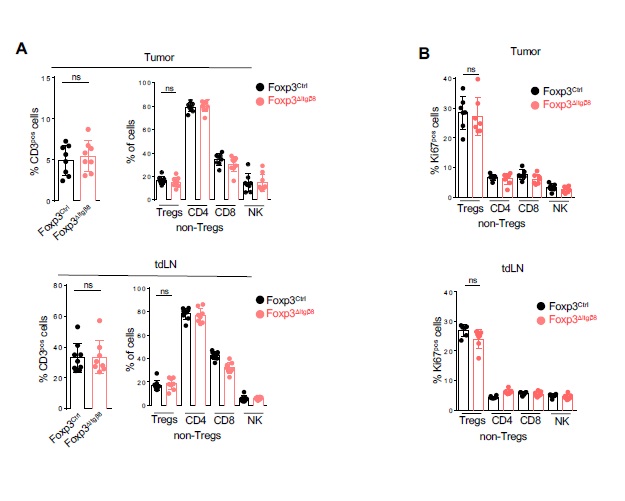
To confirm this scenario, we next analyzed the immune compartment of tumors and that of their draining lymph nodes (tdLN). Interestingly, the proportion of Natural Killers (NK) cells and T cells, including Tregs, were similar in both TME and tdLN between Foxp3ΔItgβ8 mice and Foxp3Ctrl animals (Figure S1A). In line with this observation, the proliferative status of T cells and NK cells was similar between Foxp3ΔItgβ8 mice control animals in both tdLN and TME (Figure S1B). Moreover, the deprivation of Itgb8 on Tregs failed to affect the distribution of T lymphocytes within the TME (data not shown). Thus, we ruled-out a specific role of the Itgβ8pos Tregs in controlling proliferation, recruiting of effector immune T cells into the TME as well as T cell priming in tdLN. However, the inhibition of tumor-growth observed in Foxp3ΔItgβ8 mice was completely lost when animals were depleted of their CD8pos T lymphocytes (Fig. 3A-B).
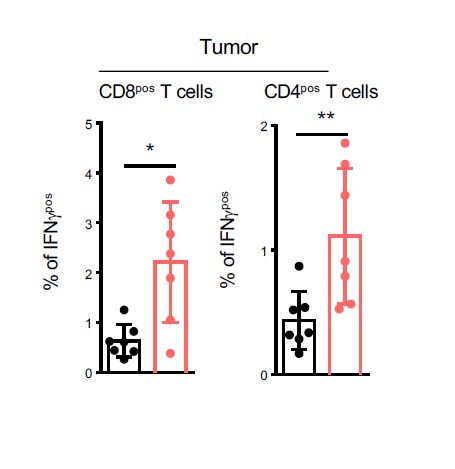
Thus, altogether these observations suggested that Itgβ8pos Tregs exert their pro-tumoral effects by impairing the anti-tumor functions of CD8pos T lymphocytes. In agreement with this assumption, we observed that CD8pos T lymphocytes of the TME of Foxp3ΔItgβ8 mice exhibited higher cytotoxic functions based on the production of granzyme B cytotoxic granules (GzB) in association with the surface expression of CD107 (Lamp1) compared to Foxp3Ctrl animals (Fig. 3C). Production of IFN-γ was also exacerbated in tumor infiltrating in both CD4Pos T cells and CD8pos T cells from Foxp3ΔItgβ8 mice compared to Foxp3Ctrl animals (Figure S2).
Supporting the exacerbated cytotoxic features of CD8pos T lymphocytes in the TME of Foxp3ΔItgβ8 mice, as well as the control of tumor growth in these animals, histology analysis showed higher numbers of apoptotic cells in tumors from Foxp3ΔItgβ8 mice than control animals (Fig. 3D-E). Importantly, in clear contrast to the TME, we failed to find any exacerbation of the cytotoxic phenotype of CD8pos T cells in the tdLN of Foxp3ΔItgβ8 mice (Fig. 3F). This observation, combined with the absence of systemic T effector cell activation in secondary lymphoid organs of in Foxp3ΔItgβ8 mice 17, 18, reveals a specific role for Itgβ8pos Tregs in the repression of the cytotoxic functions of CD8pos T lymphocytes selectively in the TME.
Altogether, these data identify Itgβ8 as a key mediator of Treg induced suppression of the anti-tumor cytotoxic function of CD8pos T cells present in the TME with direct consequences on tumor progression.
Itgβ8 expression on Tregs promotes TGF-β signaling controlling effector tumor T cells
Given the role of αvβ8 in TGF-β activation 11, and the unique ability of the Itgβ8pos Treg subset to activate TGF-β1 compared to Itgβ8neg Tregs (figure 1Ι), we next assessed whether the repression of CD8pos T cell cytotoxic functions in the TME of Foxp3ΔItgβ8 mice was due to an increase of the TGF-β signaling in the effector cells in the tumor. This assumption was even more motivated by the fact that, we found that the percentage of T cells with high activation of TGF-β signaling pathway, monitored by the phosphorylation of SMAD2-3, was halved in the TME of Foxp3ΔItgβ8 mice compared to Foxp3Ctrl animals (Fig. 4A). Notably, in contrast to the TME, and in line with the absence of T cell over activation in tdLN of Foxp3ΔItgβ8 mice the levels of phosphorylation of SMAD2-3 in T lymphocytes from tdLN were similar between Foxp3ΔItgβ8 mice and Foxp3Ctrl animals (Fig. 4B). Hence, Itgβ8pos Tregs are responsible of the increase of TGF-b signaling in T cells present in the TME.
In order to confirm that the exacerbated cytotoxic features of CD8pos T cells in the TME of Foxp3ΔItgβ8 mice were directly linked to the increase TGF-β signaling in effector T cells by Itgβ8pos Tregs, we developed genetic approaches allowing to sustain high levels of TGF-β signaling activation in effector T cells. The T cell compartment of CD3ε deficient mice was reconstituted with purified Tregs from either Foxp3Ctrl mice or Foxp3ΔItgβ8 mice and Foxp3neg T cells expressing either a constitutively active (CA) form (TGFβRICA) or the unmodified form of TGFβRI (TGFβRIWT) 19(Fig. 4C). In TGFβRICA-expressing T cells, the TGF-β signaling pathway remains activated even in the absence of bio-active source of TGF-β in their micro-environment as we previously described it 19, 20. Similarly to data illustrated in Fig. 3C, we observed that the absence of Itgβ8 expression in Tregs (TregΔItgβ8) increased the cytotoxic features of transferred wild type CD8pos T cells. In contrast, the maintenance of TGF−β signaling in effector T cells was sufficient to completely prevent the over-activation of their cytotoxic program as well as the repression of tumor growth we routinely observed in the absence of Itgβ8 expression on Tregs (Fig. 4D-E). Thus, within the TME, Itgβ8 expression on Tregs increases the levels of TGF-β signaling activation in effector T lymphocytes which is sufficient to repress their cytotoxic functions.
Activation of cancer-cell-produced TGF-β1 by Itgβ8posTregs leads to tumor CD8 T cell loss of function
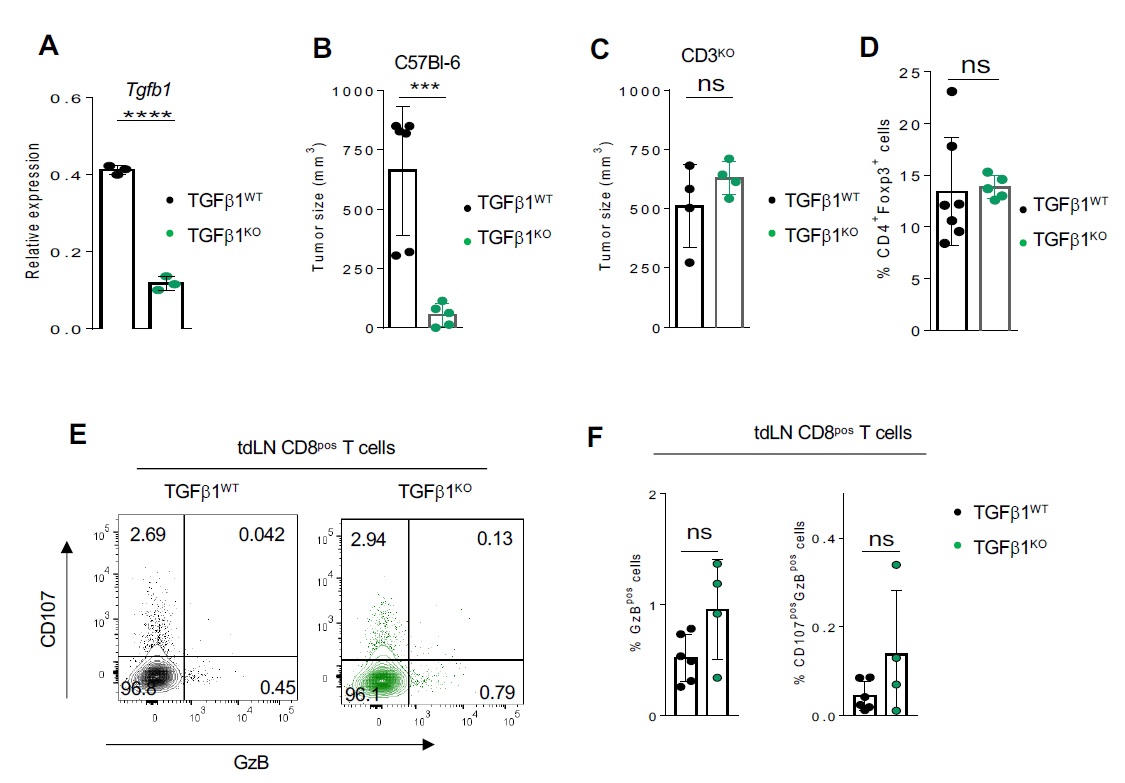
Our aforementioned data, combined with inability of Itgb8 expression to modulate Tgf-b1 expression in Tregs (Fig. 1I) and the minor role of TGF-β1-produced by Tregs in the control of the effector T cell functions in the TME 12, strongly suggest that Itgb8pos Tregs could contribute to the activation of TGF-b1 produced by other cells of the TME. As LAP reflects the inactive form of TGF-β, we evaluated the presence of LAP within the TME either in the presence or in the absence of Itgβ8 in Tregs. Strikingly, the classic fibrillar staining of the large latent complex was 2–3 times increased in the TME of Foxp3ΔItgβ8 mice compared to Foxp3Ctrl animals (Fig. 5A-B). In order to address, the source of inactive TGF-b1 which accumulate in the TME of Foxp3ΔItgβ8 mice, we selective ablated tgf-b1 in cancer cells regarded as high producer cells of TGF-b1 (TGF-β1KO) in the TME 23 (Figure S3A). The accumulation of inactive form of TGF-β1 was lost in the TME of TGF-β1KO cancer cells (Fig. 5C). Of note, the absence of TGF-b1 production by cancer cells strongly impaired the tumor growth in wild-type mice but not in T cell deficient animals (CD3KO) (Figure S3B C). Confirming the importance of TGF-b1 produced by cancer cells in the control of T cell anti-tumor immune response, the cytotoxic functions of CD8 T cells from the TME of TGF-β1KO cancer cells were 2–3 times exacerbated TGF-β1 sufficient cancer cells (Fig. 5D-E). Importantly the production of TGF-b1 by cancer cells had no significant impact Treg homeostasis (Figure S3D) and T cell activation in the tdLN (Figure S3E-F). Thus, Itgb8 expression by Tregs contributes to the activation TGF-b1 produced by cancer cells in the TME, with direct consequences on the repression the cytotoxic functions of CD8pos T cells present in the TME and thus on tumor immune escape .
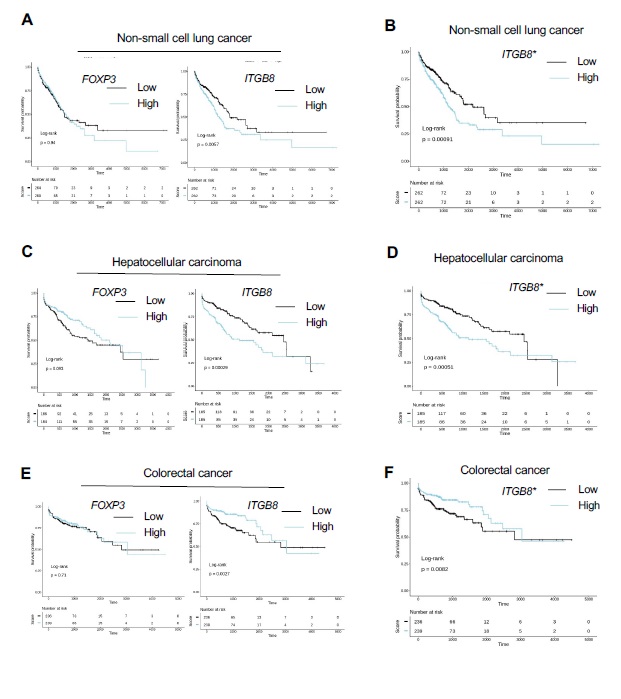
Itgβ8 expression on tumor infiltrating T cells is associated with poor patient survival and CD8 T cell activation
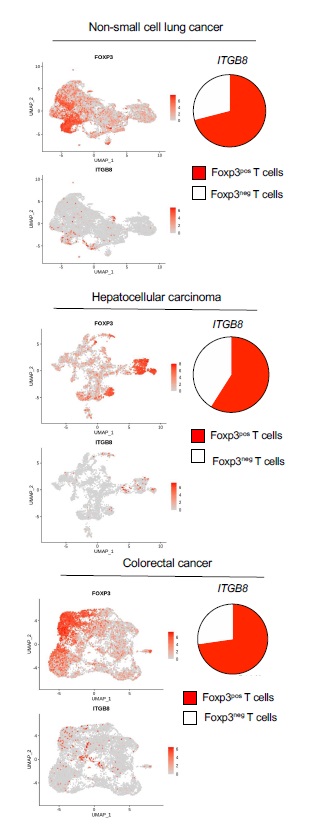
We next analyzed the relevance of our data in mice to the human pathology particularly in melanoma patients. First, we confirmed that human T cells expressed ITGb8 in the TME by analyzing single-cell mRNAseq, and reported that ITGB8 expression was prevalent in the Foxp3pos compartment of the TME of various tumor types, with 65–70% of Itgβ8pos T cells being Foxp3pos T cells (Figure S4). We then made use of publicly available sets of single cell-sequencing analysis data and obtained a specific gene-expression signature of Itgβ8pos T cells infiltrating the tumors, allowing us to perform multivariable survival analysis. We analyzed 358 patients bearing melanoma and revealed that high ITGB8 score in tumor infiltrating T cells was associated with poor survival (Fig. 6A). Of note the poor survival prognostic associated to presence of Itb8 Tregs was confirmed in other tumor types except in colorectal cancer (Figure S5) The better survival prognostic observed in colorectal patients with high ITGB8 score in Tregs from the TME was in agreement with the ability of Itgβ8pos Tregs to repress established chronic intestinal inflammation in mice 18 which was largely depicted to promote colorectal cancer progression 21. Interestingly, our analysis confirmed that FOXP3 expression alone in the T cells of TME was not sufficient to predict patient prognosis in any tumor types as previously showed 22 (Figure S5). Of note, given that ITGB8 expression was reported to be increased on activated human Tregs 18, we also removed the gene signature of activated Tregs in the ITGB8 Treg signature and obtained similar survival prognostics as with the ITGB8 Treg total gene-signature for all the tumor types we analyzed (Figure S5). In line with poor survival associated with the presence of Itgb8 Tregs in the TME of patients, we observed that the expression of ITGB8 Treg signature in the TME was inversely corelated with the activation of CD8 T cells present in the same TME (Fig. 6B). Thus, these data suggest that ITGB8 expression in Tregs present in the TME might be useful as predictor of poor patient survival and activation of CD8 T cells in tumors. Moreover combined with our analysis in mice, the aforementioned observations suggest that neutralizing Itgb8 ability to activate TGF-b in patient tumors could be associated with stronger CD8 T cells activation in the TME.
Neutralization of Itgβ8 exacerbates cytotoxic T cell function in TME of patients
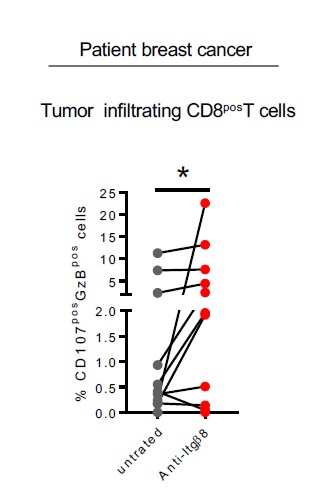
Finally, we assessed whether neutralizing Itgb8 ability to activate TGF-b in patient tumors could affect effector t cells ability to respond to TGF-b and develop efficient anti-tumor response in the TME. To this end, we used an ex-vivo culture approach in which two serial sections of live tumor were cultured either in the presence or in the absence of neutralizing anti-Itgβ8 antibody (Fig. 7A). This technique allowed us to address the effects of the anti-Itgb8 antibody on same TME of given same patient in which the immune system compartment and its interactions with the tumor tissues were conserved. After treatment, CD8pos T cells from the tumors were analyzed by flow cytometry (Fig. 7B). We first monitored the effects of the anti-Itgb8 antibody treatment on TGF-b signaling in patient melanoma. In response to anti-Itgβ8 antibody, we observed 30–50% of reduction in phosphorylation of SMAD2/3 in CD8pos T cells from TME demonstrating that neutralizing Itgβ8 in the human tumors affects the levels of TGF-β signaling in CD8pos T cells infiltrating the TME (Fig. 7C-D). Strikingly, we also observed a 2–5 fold-increase of cytotoxic features of CD8pos T cells present in the TME in the majority of the melanoma after anti-Itgβ8 antibody treatment compared to untreated condition (Fig. 7E-F). Of note, similar observations were made in breast cancers in response to neutralizing anti-Itgβ8 antibody treatment (Figure S6). Thus, neutralizing Itgβ8 is sufficient to impair TGF-β signaling in CD8pos T lymphocytes infiltrating human tumors and boost their cytotoxic functions, opening the path towards clinical applications based on Itgb8 targeting in cancer.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}