Clinical background of patient 89
The selected CRC patient is a 58-year-old woman who does not consume alcohol or tobacco, diagnosed with CRC in November 2020 in HUAC (Spain). This woman underwent a colon resection by laparoscopy in December 2020 (Fig. 1A). Adjuvant chemotherapy treatment was not administered before resection. Primary tumor, an intestinal ulcerated adenocarcinoma, was located on right colon (cecum) and classified as stage IIA (T3N0). No vascular and/or perineural invasion was detected. Furthermore, three tubular low grade dysplasia adenomas were located throughout the right colon section. Histopathological study revealed an intestinal adenocarcinoma with low grade tumor budding. Primary tumor presented a driver mutation in KRAS gene (G12X). No alterations in expression of DNA mismatch repair proteins (coded by MLH1, MSH2, MSH6 and PMS2 genes) or mutations in NRAS, BRAF genes were detected. No microsatellite instability was found. Moreover, in February 2022 this patient was diagnosed with liver metastasis (VII segment) and surgical resection was performed in the same hospital. Molecular analyses in metastasis biopsy did not detect any mutations in KRAS, NRAS or BRAF genes or microsatellite instability. The expression of mismatch repair proteins was also preserved. No lymph node affectation was detected in this patient even before any neoplastic lesion in the liver was noticed. All these clinical data were obtained from the Pathological Anatomy and General and Digestive Surgery Services of CHUAC.
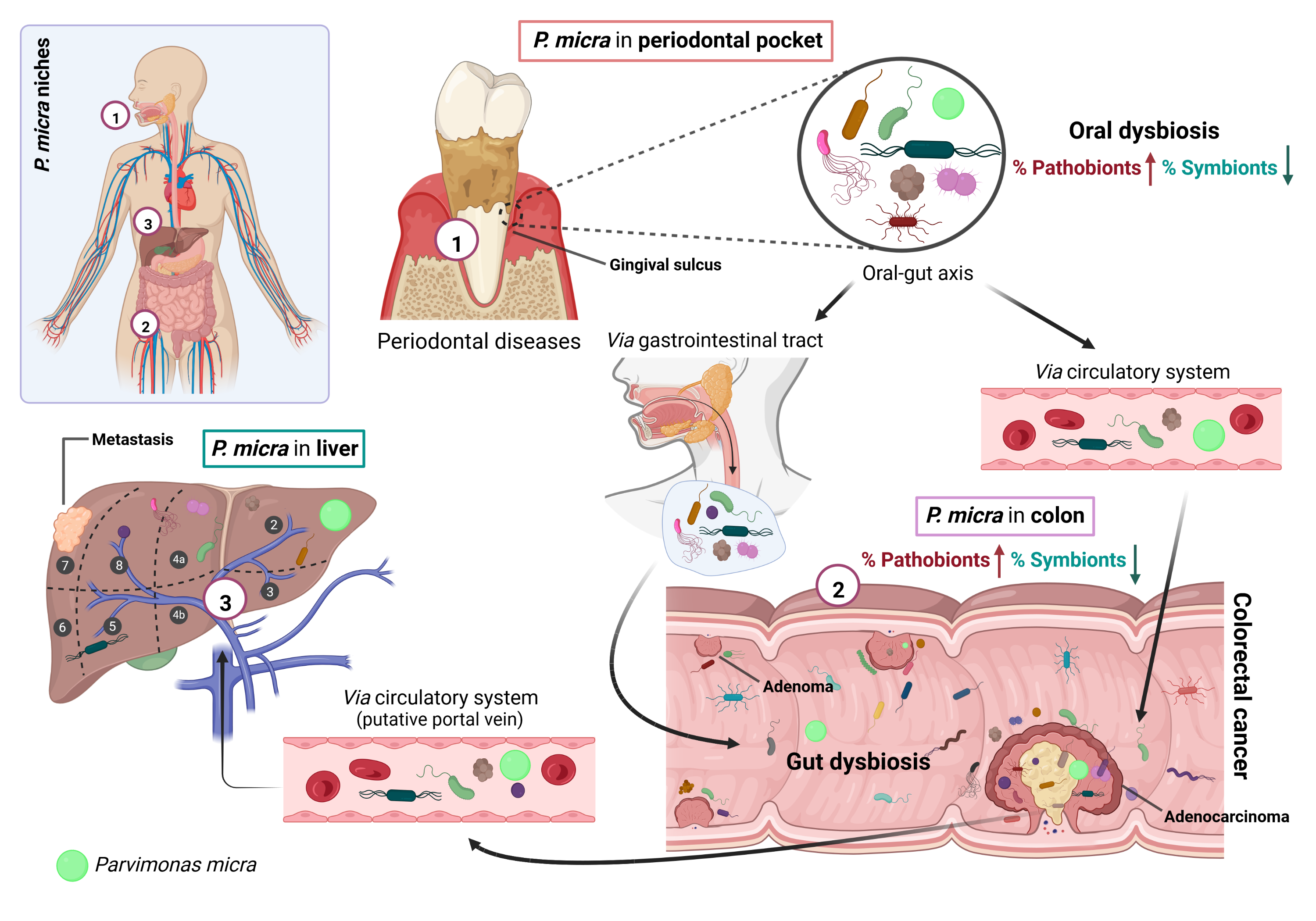
An intraoral and extraoral examination of the patient was performed at Pardiñas Dental Clinic (A Coruña, Spain). Clinical measurements of probing depth, clinical attachment level, and bleeding on probing were recorded at six points around each tooth. Miller's mobility index was recorded for each tooth. A Silness Loe score of 0.3 for bleeding on probing and a CPO index for dental caries (tooth cavities) of 7 were obtained, indicating that the patient had a high degree of tooth decay and tooth loss. The Cone Beam Computed Tomography (CBCT) from the patient’s upper and lower jaw revealed an advanced degree of bone loss, the furcation involvement of teeth numbers 16, 26, 27 and 38, and the absence of teeth numbers 18, 17, 28, 35, 36, 37, 46, 47 and 48. Figure 2 shows the generalized horizontal bone loss and the high-grade furcation involvement detected in the oral cavity of P89 after oral exploration. Based on these data, a diagnosis of periodontitis in stage IVB was made, according to the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions [87].
In-depth analysis of P. micra genomes from strains isolated from patient 89
Based on a whole genome alignment phylogenetic tree (Fig. 3), which includes eleven P. micra genomes described in the present study and twelve P. micra genomes obtained from NCBI database (Table 1), it can be concluded that isolates from P89 belong to a very well differentiated group, distinguishing themselves from other P. micra isolates obtained in this study and from the rest of P. micra genomes (Fig. 3).
Furthermore, pangenome clustering revealed differences in the genes shared between strains, including differences between gingival and adenocarcinoma isolates of P89 (Fig. 4).
Due to these findings, an in-depth genome comparison of Parvimonas genomes from P89 was conducted. A total of 2120 non-synonymous mutations were found (Additional file; Table S1), including 1298 SNPs, 745 complex mutations, 40 deletions and 37 insertions, between oral PM89KC-G isolates 1 and 2, which were virtually identical, and the tumor PM89KC-AC-1 isolate. Inside these mutations, a total of 1603 genes were affected.
When studying presence and absence of genes between oral and tumor isolates from P89, some differences were detected (Tables 2 and 3). The biggest difference corresponded to a fragment of 25728 bp containing a group of 23 genes found in both oral isolates and absent in the tumor strain (Table 2). Additionally, an identical transposase element repeated at several different genome locations was present in PM89KC-AC-1 and absent in the oral isolates PM89KC-G 1 and 2 (Table 3).
Table 2
Missing genes in P. micra PM89KC-AC-1 isolate when compared to gingival PM89KC-G 1/ 2 isolates.
| Product | Average length (bp) | Cluster | Locus Tag |
| PM89KC_G_1 | PM89KC_G_2 |
| TetR/AcrR family transcriptional regulator* | 573 | 24 | NM219_06315 | NM220_06315 |
| ABC transporter ATP-binding protein/permease* | 1740 | 24 | NM219_06310 | NM220_06310 |
| ABC transporter ATP-binding protein/permease* | 1710 | 24 | NM219_06305 | NM220_06305 |
| ATP-binding cassette domain-containing protein* | 1518 | 24 | NM219_06300 | NM220_06300 |
| Energy-coupling factor transporter transmembrane protein EcfT* | 705 | 24 | NM219_06295 | NM220_06295 |
| MptD family putative ECF transporter S component* | 585 | 24 | NM219_06290 | NM220_06290 |
| Sigma-70 family RNA polymerase sigma factor* | 411 | 24 | NM219_06285 | NM220_06285 |
| Hypothetical protein* | 162 | 24 | NM219_06280 | NM220_06280 |
| Hypothetical protein* | 567 | 24 | NM219_06270 | NM220_06270 |
| Exonuclease domain-containing protein* | 1872 | 24 | NM219_06265 | NM220_06265 |
| Hypothetical protein* | 1935 | 24 | NM219_06260 | NM220_06260 |
| Hypothetical protein* | 885 | 24 | NM219_06255 | NM220_06255 |
| Hypothetical protein* | 618 | 24 | NM219_06250 | NM220_06250 |
| Hypothetical protein* | 1113 | 24 | NM219_06245 | NM220_06245 |
| Hypothetical protein* | 387 | 24 | NM219_06240 | NM220_06240 |
| InlB B-repeat-containing protein* | 1809 | 24 | NM219_06235 | NM220_06235 |
| Putative ABC transporter permease* | 717 | 24 | NM219_06230 | NM220_06230 |
| Branched-chain amino acid transport system II carrier protein* | 1287 | 24 | NM219_06225 | NM220_06225 |
| O-acetylhomoserine aminocarboxypropyltransferase/cysteine synthase* | 1290 | 24 | NM219_06220 | NM220_06220 |
| DKNYY domain-containing protein* | 1479 | 24 | NM219_06215 | NM220_06215 |
| LPXTG cell wall anchor domain-containing protein* | 2193 | 24 | NM219_06210 | NM220_06210 |
| MBL fold metallo-hydrolase* | 810 | 24 | NM219_06205 | NM220_06205 |
| Aldehyde dehydrogenase* | 1362 | 24 | NM219_06200 | NM220_06200 |
| Hypothetical protein | 324 | 25 | NM219_00070 | NM220_00070 |
| IS200/IS605 family transposase | 459 | 25 | NM219_00075 | NM220_00075 |
| Hypothetical protein | 1104 | 25 | NM219_00080 | NM220_00080 |
| DUF1307 domain-containing protein | 486 | 26 | NM219_00915 | NM220_00915 |
| DUF1307 domain-containing protein | 477 | 26 | NM219_00920 | NM220_00920 |
| DUF2087 domain-containing protein | 270 | 27 | NM219_01675 | NM220_01675 |
| GNAT family N-acetyltransferase | 480 | 27 | NM219_01680 | NM220_01680 |
| ABC transporter permease | 1263 | 28 | NM219_06715 | NM220_06715 |
| Hypothetical protein | 636 | 29 | NM219_03355 | NM220_03355 |
| Hypothetical protein | 372 | 30 | NM219_00160 | NM220_00160 |
| Hypothetical protein | 183 | 31 | NM219_03780 | NM220_03780 |
| ABC transporter ATP-binding protein/permease | 1611 | 32 | NM219_01745 | NM220_01745 |
| Hypothetical protein | 1638 | 33 | NM219_06615 | NM220_06615 |
| Hypothetical protein | 198 | 34 | NM219_00320 | NM220_00320 |
| Hypothetical protein | 432 | 35 | NM219_00645 | NM220_00645 |
| Hypothetical protein | 147 | 36 | NM219_03720 | NM220_03720 |
| CPBP family intramembrane metalloprotease | 678 | 37 | NM219_06015 | NM220_06015 |
| GntR family transcriptional regulator | 150 | 38 | NM219_02485 | NM220_02485 |
| Hypothetical protein | 180 | 39 | NM219_07000 | NM220_07000 |
| Hypothetical protein | 384 | 40 | NM219_06370 | NM220_06370 |
| Asterisks (*) indicates a group of 23 genes (25728 bp) belonging to cluster 24 present in the oral isolates (PM89KC-G 1 and 2) and absent in the tumor isolate (PM89KC-AC-1). All listed genes were 100% identical between the two gingival isolates. |
Table 3
Gained genes in the adenocarcinoma PM89KC-AC-1 isolate when compared to the PM89KC-G 1 and 2 gingival isolates.
| Consensus product | Average length (bp) | Cluster | Locus tag |
| Recombinase family protein | 621 | 41 | NM221_06205 |
| SpaA isopeptide-forming pilin-related protein | 2271 | 41 | NM221_06200 |
| Pseudouridine-5'-phosphate glycosidase | 546 | 42 | NM221_06515 |
| ABC transporter ATP-binding protein/permease | 1560 | 42 | NM221_06510 |
| EXLDI protein | 369 | 43 | NM221_01675 |
| Transposon-encoded TnpW family protein | 231 | 43 | NM221_01680 |
| Restriction endonuclease subunit S | 1242 | 44 | NM221_00825 |
| Hypothetical protein | 435 | 45 | NM221_00630 |
| Rhodanese-like domain-containing protein | 1083 | 46 | NM221_01690 |
| Hypothetical protein | 1017 | 47 | NM221_05350 |
| IS630 family transposase* | 1194 | 48 | NM221_00050; NM221_00875; NM221_01125; NM221_01455; NM221_02510; NM221_06115; NM221_07165; NM221_07365 |
| ABC transporter ATP-binding protein/permease | 1617 | 49 | NM221_06020 |
| Hypothetical protein | 360 | 50 | NM221_06500 |
| Hypothetical protein | 2496 | 51 | NM221_01205 |
| ParB/RepB/Spo0J family partition protein | 606 | 52 | NM221_07790 |
| Relaxase/mobilization nuclease domain-containing protein | 1332 | 53 | NM221_06215 |
| Asterisks (*) tag an identical transposase element repeated at several different genomic locations present in the tumor isolate (PM89KC-AC-1) and absent in the gingival isolates (PM89KC-G 1 and 2). |
Synteny analysis also revealed loss and gain of genes, but more interestingly, a specific cross-shaped structure in PM89KC isolates (Fig. 5). This “genomic cross” was composed of a repeat region of 30 genes (pairwise identity of ~ 80%) flanking the ~ 600 Kbp and ~ 800 Kbp positions (Additional file; Table S2), present in all P89 isolates.
These repeats correspond to a shared region in two very similar prophages, where manual inspection revealed multiple genes involved in genomic mobility and recombination (recombinase, replication initiator protein, DNA binding protein, conjugal transfer protein, topoisomerase, helicase, relaxases, relaxosome proteins, helix-turn-helix transcriptional regulator and sigma 70 family RNA polymerase sigma factor), but none involving capsid or tail virus formation. These prophages have been detected in other P. micra isolates in different positions, although usually not duplicated. When the whole left prophage of PM89KC-AC-1 (~ 42 Kbp, position 564183...607520) was compared against OVD and the GPD (minimum identity ≥ 50%), good hits (e-value of 0 and bitscore over 3000) were found with a median identity of 89.226 ± 4.65% in OVD. The top hits belonged to a 42456 bp unclassified prophage (median identity of 89.77 ± 2.35%), which appears under different hosts such as Oribacterium, Fusobacterium, Streptococcus, Prevotella, Anaerosphaera and Peptoanaerobacter. The search did not return hits in the GPD. Additionally, putative virulence factors were detected in the prophages, including: an ABC transporter ATP-binding protein and a Type IV secretion system (including associated proteins PcfB and PrgI). Other relevant proteins found were a NlpC/P60 family protein in the left prophage and a CHAP domain containing protein in the right prophage. These prophages shared a large proportion of genes, but the left one had several extra proteins (Additional file; Table S2).
If KCOM 1037 strain is taken as a reference, the left prophage has been inserted into a CRISPR array of a type III-B CRISPR-Cas system in all P89 isolates (Additional file; Figure S2), separating the Cas proteins (CRISPR associated proteins) from the CRISPR array. However, the PM89KC-G and PM89KC-AC-1 isolates differ in the number and length of CRISPR arrays on both sides of the prophage. Isolate PM89KC-G-1 presents two CRISPR arrays after the prophage. The first one (G-A), of 298 bp, is located at position 598786, and the second one (G-B), of 423 bp, at position 601696. Isolate PM89KC-AC-1 presents these two arrays after the prophage: the first one (AC-B), of 2213 bp, located at position 607555, and second one (AC-C), of 423 bp, at position 612379 and an extra array (AC-A, position 563270, 749 bp) before the prophage (Additional file; Figure S2). Arrays G-A and G-B from PM89KC-G-1 correspond to arrays AC-B and AC-C in PM89KC-AC-1. The repeats in all these arrays share the same core structure (Additional file; Table S3). All spacers inside array G-B from PM89KC-G-1 are exactly identical to the ones in array AC-C in PM89KC-AC-1 and the last 3 spacers from AC-B are the ones in G-A. Furthermore, when searching for these spacers in other P. micra isolates, none contained the exact same ones, and were only shared in P89. If the subgingival isolates are taken as reference origin, the adenocarcinoma isolate could have increased the size of array G-A from 3 spacers to 32 and gained the extra array AC-A with 11 spacers (total net gain of 40 spacers). The first two spacers of AC-A on the left of the prophage for PM89KC-AC-1 were also detected before the prophage in PM89KC-G (G-Res), further supporting the common origin of these isolates. However, when comparing spacers from EYE_30 and PM79KC-G-1, many were identical, which indicates that sharing spacers across isolates is not uncommon and cannot be used as definitive proof. When the new spacers of PM89KC-AC-1 were searched against OVD and GPD, a couple in array AC-B matched unclassified phages in genus Coprococcus and Tyzzerella (both from family Lachnospiraceae) in the GPD. The rest matched oral phages in Parvimonas, Streptococcus and Fusobacterium in OVD or intestinal phages in Parvimonas in GPD. Finally, a similar insertion and duplication of this prophage has been seen in other P. micra such as PM102KC-G-1, but in a different CRISPR-Cas system (Type CAS-II-A/CAS-III-A) with different spacers and repeated sequences (Additional file; Table S3).
Potential virulence factors were analyzed in gingival and adenocarcinoma isolates of P89 using VFDB (Additional file; Tables S4 and S5). Most virulence factors were shared between PM89KC strains including multiple iron scavenging and transport proteins, type III, IV and VII secretion systems, colibactin toxins, neutrophil activating proteins, tissue adhesins, peptidases and biofilm regulators.
Interestingly, even though these isolates were > 99% identical, the phenotype of the gingival isolates extracted from P89 was considerably different to the adenocarcinoma isolate. The gingival P. micra colonies were completely white and compact with well-defined borders whereas the adenocarcinoma P. micra isolate colonies were translucent, brighter and had undefined borders. No hemolytic activity was observed for any strain (Additional file; Figure S3).
It is important to note that after the liver metastasis diagnosis for P89, our team collected, during the laparoscopy surgery, metastatic and non-metastatic liver samples in order to culture both specimens and to perform a 16S rRNA metabarcoding analysis. However, efforts to isolate P. micra in the liver were unsuccessful.
Thus, given that the above results suggest a possible same origin of the adenocarcinoma and the gingival P. micra isolates of P89, we decided to focus on a complete 16S metabarcoding and metatranscriptomic analysis of the different samples from this patient.
Microbiome of patient 89
To characterize the microbiome of the CRC P89 we performed a deep analysis of the bacterial 16S rDNA in feces, saliva, subgingival fluid and non-neoplastic, transition and adenocarcinoma tissues as well as in metastatic and non-neoplastic liver regions (Fig. 1).
The microbiome analysis of feces (M89-F) revealed that Faecalibacterium (26.16%), Enterococcaceae bacterium RF39 (20.73%), Eubacterium coprostanoligenes group (9.02%), Collinsella (7.71%) Ruminococcus gnavus group (7.61%), Lachnospiraceae (2.86%) and Bacteroides (2.47%) were the most abundant bacteria (Fig. 6).
Additionally, the taxonomic assignment of the saliva sample (M89-S) sequences showed that the most common genera were Streptococcus (38.04%), Neisseria (12.36%), Capnocytophaga (6.83%) and Granulicatella (4.97%) followed by Gemella (4.40%), Fusobacterium (4.33%) and Porphyromonas (4.24%). When gingival crevicular fluid sample (M89-G) was analyzed, the genus Fusobacterium (26.30%), Porphyromonas (24.73%), Prevotella (9.15%), Dialister (9.05%), Tannerella (4.94%), Parvimonas (2.83%) and a member of the Peptostreptococcaceae family (Peptostreptococcaceae bacterium oral taxon 113 str. W5053 at 3.93%) were found as the most abundant bacteria (Fig. 6). In agreement with the bad periodontal health of the patient, two bacteria species pertaining to the red complex group, P. gingivalis and Tannerella forsythia, were detected in both M89-S and M89-G samples, as well as other important periodontal pathogen species (some of them belonging to orange and green complex groups) such as Porphyromonas endodontalis, Prevotella intermedia, Dialister pneumosintes, Eubacterium nodatum or Mogibacterium timidum (Fig. 6).
Fresh tissue adenocarcinoma sample (M89-FT-Ac) collected during the laparoscopy surgery was also analyzed. The tumor microbiome, mostly shaped by anaerobic bacteria, was composed of five major genera: Bacteroides (31.69%), Fusobacterium (25.62%), Peptostreptococcus (6.49%), Prevotella (5.17%) and Hungatella (4.16%) (Fig. 6). In addition, transition tissue (M89-FT-Tr) and non-neoplastic colon mucosa tissue (M89-FT-NC) were collected in order to understand the microbe evolution in the colon mucosa. The analysis of the PM89-FT-Tr sample revealed that its microbiome was mainly composed by Bacteroides (45.80%), Faecalibacterium (13.92%), Lachnospiraceae (9.03%), Ruminococcus gnavus group (4.51%), Ruminococcus torques group (2.02%), Barnesiella (1.91%), Faecalitalea (1.64%), Peptostreptococcus (1.49%) and Prevotella (1.45%). Besides, the M89-FT-NC microbiome was composed by similar bacteria but with a significantly different abundance when compared to transition and adenocarcinoma tissues (Fig. 6). For example, the bacteria B. fragilis, over-represented in Ac sample (25.17%), showed low abundance in normal colon tissue sample (1.66%) and also in M89-FT-Tr tissue (1.72%). This tendency was also observed for other microorganisms such as Peptostreptococcus anaerobious, which was over-represented in adenocarcinoma (5.58%) and low abundant in transition (0.70%) and normal (0.82%) colon tissues. P. intermedia was the third most abundant species identified in adenocarcinoma (5.17%) but it was less frequent in transition (1.45%) and in normal (1.19%) mucosa tissues. This happened also for C. showae and Streptococcus agalactiae, which showed higher abundance in M89-FT-Ac sample (2.96% and 0.99%, respectively), while their presence in M89-FT-Tr tissue (0.08% and 0.11%, respectively) or in M89-FT-NC tissue (0.05% and 0.09%, respectively) was very low. In contrast, the microorganism B. dorei appeared with higher relative abundance in M89-FT-Tr tissue (21.73%) or in M89-FT-NC tissue (14.00%) compared to M89-FT-Ac (3.06%) (Fig. 6).
The microbiome analysis performed in the liver sample revealed that Streptococcus (20.91%), Pseudomonas (15.46%), Haemophilus (10.61%), Staphylococcus (6.75%) and Veillonella (5.54%) were the main genera found in the non-neoplastic adjacent tissue (M89-FT-NL) while in the metastatic region (M89-FT-MetL), Pseudomonas (33.74%), Streptococcus (10.51%), Bacteroides (6.63%) and Rothia (4.34%) were the most represented genera.
It is important to note that periodontal pathogens genera, such as Capnocytophaga, Parvimonas, Prevotella and Eubacterium, were detected, in some cases with low abundance, in liver samples of P89.
Parvimonas was found in colon in adenocarcinoma, non-neoplastic and transition tissues (1.24%, 0.65% and 0.27%, respectively) being enriched in cancerous tissue. Parvimonas was also present in feces (1.00%), saliva (1.34%) and subgingival crevicular sample (2.83%). In the liver sample Parvimonas was detected with a relative abundance of 3.18%. P. micra was grown after culturing gingival and colorectal adenocarcinoma but no P. micra colonies were obtained after culturing the liver tissue.
As commented before, other typical oral pathogens were detected in all types of samples, being the most abundant Fusobacterium, Prevotella, Campylobacter and Dialister. Figure 6B shows bacteria shared between gingival and adenocarcinoma samples.
FFPE samples revealed a similar bacterial composition (Fig. 7). Typical oral microbes were detected in those FFPE samples, as well as in non-paraffin embedded samples, such as Prevotella (PT-NC: 0.87%; PT-A: 1.60%; PT-Ac: 1.08%), Fusobacterium (PT-NC: 0.37%; PT-A: 0.70%; PT-Ac: 26.50%), Dialister (PT-NC: 0.01%; PT-Ac: 0.04%), Actinomyces (PT-NC: 0.16%; PT-A: 0.31%; PT-Ac: 0.20%), Gemella (PT-NC: 0.15%; PT-A: 1.03%; PT-Ac: 0.22%) and Parvimonas (PT-NC: 0.02%; PT-A: 0.13%; PT-Ac: 0.01%).
Metatranscriptomics of tumor tissues
In order to evaluate bacterial activity in the tumor tissue of P89, where Parvimonas was isolated, a metatranscriptomic analysis was performed from colon tissues (Fig. 1). In particular, we analyzed the adenocarcinoma tissue (T89-FT-Ac), as well as non-neoplastic colon mucosa tissue (T89-FT-NC) and transition tissue (interface between non-neoplastic and adenocarcinoma region, T89-FT-Tr).
At the species taxonomic level, a higher richness and diversity were observed in the non-neoplastic tissue compared to the adenocarcinoma sample (Table S6). Microbial activity profile was different at the species level when these samples were compared (Fig. 8A). The active microbiota in the non-neoplastic colon tissue (T89-FT-NC) and colon transition tissue (T89-FT-Tr) from P89 was dominated by B. dorei (FT-NC: 25.55%, FT-Tr: 39.53%, FT-Ac: 5.68%), Faecalibacterium prausnitzii (FT-NC: 6.06%, FT-Tr: 6.94%, FT-Ac: 0.87%), Faecalicatena gnavus (FT-NC: 8.35%, FT-Tr: 3.29%, FT-Ac: 1.27%) and Bacteroides thetaiotaomicron (FT-NC: 2.83%, FT-Tr: 6.72%, FT-Ac: 0.59%). In contrast, B. fragilis (FT-N: 1.65%, FT-Tr: 2.74%, FT-Ac: 12.94%), Peptostreptococcus anaerobius (FT-NC: 0.73%, FT-Tr: 0.78%, FT-Ac: 10.29%) and Fusobacterium polymorphum (FT-N: 0.3%, FT-Tr: 0.23%, FT-Ac: 8.96%) dominated the adenocarcinoma tissue. In addition, other oral associated species like Fusobacterium species, P. intermedia and P. micra also accounted for a higher percentage of activity in adenocarcinoma than in the non-neoplastic tissue. Parvimonas was detected by 16S rRNA gene sequencing and also metatranscriptomic analysis in the three different tissues. Moreover, Parvimonas, represented a higher percentage of transcripts (MTT) and a higher ratio of percentage of transcripts vs the relative abundance in 16S rRNA metabarcoding (16S) (MTT/16S: 1.58 in T89-FT-NC, 2.5 in T89-FT-Tr and 3.33 in T89-FT-Ac) in the adenocarcinoma tissue than in the other tissues (Fig. 9A).
Since no replicates were obtained, no statistical tests could be performed. Therefore, we considered a gene to be overexpressed when it had a mean abundance (RPKM) higher than 5 and a difference in abundance (log2[foldchange]; log2FC) > 1). According to these criteria, we found 16 genes that were overexpressed in non-neoplastic tissue and 14 genes in the adenocarcinoma (Fig. 8B).
Interestingly, among those overexpressed in the adenocarcinoma we found some stress indicators such as dps; starvation-inducible DNA-binding protein or arsR transcriptional regulator (implicated in ion homeostasis, biofilm formation, primary and secondary metabolism, response to adverse condition, and virulence). In addition, increased expression of ompW, an outer membrane protein which acts as a receptor for colicin S4 (colicins are plasmid-encoded toxic proteins produced by Escherichia coli strains), or the tcdAB, toxin A/B (pro-inflammatory and cytotoxic, causing disruption of the actin cytoskeleton and impairment of tight junctions in human intestine) were also found.
Other genes related with metabolism like different metal transporters (copA, Cu+-exporting ATPase; cbiN; cobalt/nickel transport protein), enzymes involved in carbon metabolism (ACADS; butyryl-CoA dehydrogenase, pycB; pyruvate carboxylase subunit B, gcvH; glycine cleavage system H protein), and a glutamate dehydrogenase (gudB) that allows the use of glutamate as a carbon source were also more expressed in adenocarcinoma. Meanwhile, in non-neoplastic tissue there are other several genes overexpressed that code for proteins involved in carbon metabolism (mdh, malate dehydrogenase; PGD,6-phosphogluconate dehydrogenase; G6PD glucose-6-phosphate 1-dehydrogenase), in amino-sugar metabolism (nagB), as well as two subunits of ribose transporter (rbsB, rbsC), atpE a F-type H+-transporting ATPase (used by aerobic organisms for synthesizing ATP) and SOD2 superoxide dismutase (which neutralizes toxic levels of reactive oxygen species).
Focusing on the transcriptional profile of Parvimonas in P89, a total of 808 different KEGG genes were assigned. The ones with higher expression were genes that codifies for several ribosome proteins, DNA replication proteins (hupA, ssb) transcription machinery (rpoA, nusG) and translation factors (tuf, fusA, infA) confirming that Parvimonas was transcriptionally active (Fig. 9B). Genes related to the metabolism of carbohydrates (gcvH, pflD, galE), amino acids (prdA, kbl, trxB) and proteins (nlpC, mltA, plsX) were also found. In fact, the most expressed gene by P. micra in adenocarcinoma was a probable lipoprotein (nlpC) that was also among the more overexpressed in this tissue globally. Further annotation of this gene with Pyre2 showed that only the last part of the protein (119 residues) was similar to the putative cell wall hydrolase (autolysin acd24020 catalytic domain, that belongs to the NlpC/P60 family) from Clostridium difficile (Table S7).
Regarding the potential virulence factors detected in the genomic analysis of isolates we found transcripts of 13 out of 25 genes identified (considering those with a nucleotide identity > 40%). All of them were more expressed in adenocarcinoma with the exception of clpP (more expressed in transition tissue) and LpxC-fabZ (more expressed in non-neoplastic tissue) (Fig. 9C).
{kind=link}