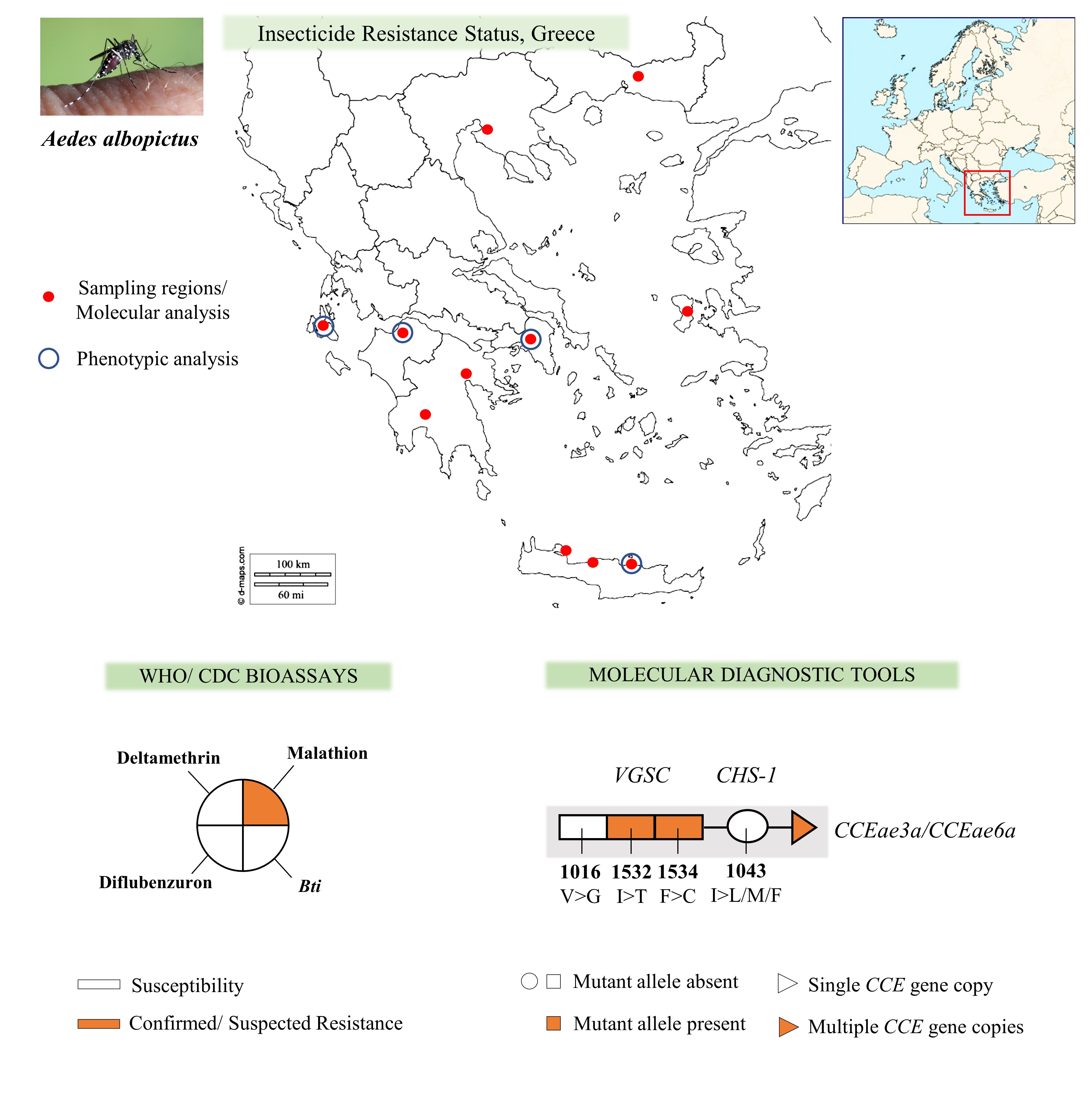
Study localities, sample collections and mosquito handling
Adult and immature stage Ae. albopictus mosquitoes were collected during the summer of 2017, 2018 and 2019, in a total of 19 urban and peri-urban localities in Greece, in the regions of Thessaloniki [33] and Rodopi (northern Greece), Attica and Argolida (central Greece), the Island of Chios (north-eastern Aegean Islands complex) [33], Patras and Kalamata (western Greece), the Island of Kefalonia (Ionian Islands complex) and Crete (Chania, Rethymno and Heraklion - southern Greece) (Fig.1, Table 1). The localities we selected to include in the analysis were chosen based on: their geographical location (in order to cover a large geographical area of Greece), the history of insecticide applications, previous insecticide resistance findings in other mosquito species and the availability of Ae. albopictus samples (mosquito collections/surveillance programmes).
Samples were collected every 2 weeks over a period of 1 or 2 months; a total of 3 to 7 collection events were conducted for each locality. Adult specimens were collected with mouth aspiration catches and CDC-light traps baited with dry ice. Larvae were sampled from natural and man-made/artificial containers with dipping collections and eggs were collected with oviposition traps (black plastic cups of 8 cm top diameter, 5 cm bottom diameter and 13 cm height; half covered with tap or rain water, with 2 wooden tongue depressors as oviposition substrate), baited with hay infusion and placed outdoors, amongst low vegetation, away from direct sunlight. Both larvae and eggs were collected from at least five different sites within each locality in order to avoid family bias and minimize the probability of including isofemale mosquitoes in the molecular analyses.
Following ovitrap collections, eggs were reared to adults in standard insectary conditions (temperature 27 ± 2 °C and relative humidity 70–80%), identified morphologically to species [34] and stored individually in absolute ethanol at 4 °C for the subsequent molecular analysis. A subgroup of eggs from the Aghios Stefanos locality (region of Attica; ovitrap collections from Aghios Stefanos were conducted in 2019, while samples analysed molecularly were collected in 2018), Kefalonia, Patras and Heraklion were reared to larvae or adults to use for susceptibility bioassays, as described below.
Genomic DNA extraction and molecular identification of mosquito species
Genomic DNA (gDNA) was extracted from individual larvae or adult mosquitoes and from pools of eggs (10 eggs per pool per locality; 3 localities), using DNAzol reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions.
Species identification was based on the PCR amplification (KAPA Taq PCR Kit; KAPA Biosystems) of the nuclear ribosomal gene spacer ITS2, following an assay that discriminates between Ae. albopictus, Ae. cretinus and Ae. aegypti, by generating PCR products of 509 bp, 385 bp and 324 bp in length, respectively, as described in Patsoula et al. [35]; the 25 µl PCR reaction contained 1 µl gDNA, 2.5 µl of 10´ DNA polymerase buffer, 2 mM MgCl2, 0.4 µM of each primer (primers 5.8S and 28S; Additional file 1: Table S1), 0.4 µM of dNTPs and 1.5 U of Taq polymerase. The applied thermal protocol was the following: initial denaturation at 94 °C for 5 min, 40 cycles ´ [denaturation at 94 °C for 1 min, primer annealing at 52 °C for 1 min, primer extension at 72 °C for 1 min] and a final extension step at 72 °C for 10 min. The PCR products were electrophoresed on a 1.5% w/v agarose gel containing ethidium bromide.
Insecticide susceptibility bioassays
Larval bioassays
Following the WHO guidelines for laboratory and field testing of mosquito larvicides [36], we examined the susceptibility of Ae. albopictus populations against two larvicides; the bacterial larvicide Bti (VectoBac12AS, Valent BioSciences LLC, Illinois, USA; 1200 ITU (international toxic units)/mg; 11.61% w/v) and the insect growth regulator DFB (DU-DIM 15SC, Arysta LifeScience, Amsterdam, The Netherlands; 15% w/v). Both insecticides were diluted in distilled water. Bioassays were performed using Ae. albopictus third-early fourth-instar larvae (F0-F1 generation), reared under standard insectary conditions (temperature 27 ± 2 °C and relative humidity 70–80%). Fifteen to 20 larvae were placed in 99 ml water, to which 1 ml of the insecticide solution was added. Control bioassays contained 100 ml water. A range of 5 to 9 concentrations were tested for each insecticide (Bti: 0.008–0.500 mg/l; DFB: 0.0004–0.0200 mg/l) in order to define a mortality range between 10 and 95% and determine the LC50 and LC95 values. Three to 4 replicates were tested for each concentration. Larval mortality was recorded after the WHO recommended exposure time for each insecticide. Moribund larvae were counted as dead [36]. LC50 and LC95 values were estimated using the log-probit analysis Polo Plus 2.0 LeOra software (LeOra Software LLC, Parma, USA). Results were compared to the values reported for susceptible-laboratory Ae. albopictus strains in other studies [37–39].
Adult bioassays
Three to five day-old, non-blood fed female mosquitoes (F1-F2 generation) were subjected to insecticide susceptibility tests against deltamethrin and malathion, following the CDC bottle bioassay guidelines [40]. A Malaysian Ae. albopictus susceptible laboratory strain was included [41]. Both insecticides were purchased as technical grade material (PESTANAL® analytical standard; Sigma-Aldrich, Darmstadt, Germany). Insecticide stock solutions were prepared in acetone and Wheaton bottles were cleaned and coated as described in the CDC guidelines. The diagnostic dose of the insecticide under evaluation was used: deltamethrin at 10 µg/bottle and malathion at 50 µg/bottle. Tests were performed using 20–25 mosquitoes per bottle. Four insecticide treated replicate bottles and at least one control bottle (coated with acetone only) were used in each experiment set. The diagnostic time for both insecticides tested was 30 min [40]. Alive and dead mosquitoes in each bottle were recorded at time intervals of 5–15 min. The insecticide susceptibility status was determined by the mortality rate at the diagnostic time, according to CDC recommendations: 98–100% mortality at the diagnostic time indicates susceptibility; 80–97% suggests the possibility of resistance that requires further confirmation; and mortality < 80% denotes resistance. In cases where mortality (between 3–5%) was recorded in the control bottles at the 2 h timepoint, mortality data were corrected using Abbott’s formula.
Genotyping of target site resistance mutations
Detection of knock-down resistance (kdr) mutations in the VGSC gene
The VGSC domain II was investigated for the presence of the V1016G mutation and domain III for mutations I1532T and F1534L/S/C via PCR and product sequencing.
The PCR (KAPA Taq PCR Kit) for domain II was carried out in 25 µl containing 1.5 µl of mixed gDNA extracted individually from 5–8 Ae. albopictus samples of the same locality, 2.5 µl of 10× DNA polymerase buffer, 0.4 µM of each primer (primers kdr2F and kdr2R; Additional file 1: Table S1), 0.4 µM of dNTPs and 1.5 U of Taq polymerase. The PCR thermal conditions were: initial denaturation at 95 °C for 5 min, 40 cycles × [denaturation at 94 °C for 30 s, primer annealing at 55 °C for 30 s, primer extension at 72 °C for 1 min] and a final extension step at 72 °C for 5 min. A small amount of the PCR products (5 µl) was electrophoresed on a 1% w/v agarose gel to verify the presence of the correct size amplicon (500 bp), and the remaining amount was purified using the Nucleospin PCR & Gel Clean-Up Kit (Macherey Nagel) and sequenced using the Sanger method (CeMIA S.A., Larissa, Greece) with primer kdr2F. Sequences were analysed using the sequence alignment editor BioEdit 7.2.5 (https://bioedit.software.informer.com/7.2/).
The PCR for domain III was carried out in 25 µl containing 1.5 µl of gDNA from Ae. albopictus individuals, 2.5 µl of 10× DNA polymerase buffer, 2 mM MgCl2, 0.3 µM of each primer (primersaegSCF7 and aegSCR7; Additional file 1: Table S1), 0.4 uM of dNTPs and 1.5 U of Taq polymerase. The thermal conditions of the PCR were: initial denaturation at 95 °C for 5 min, 40 cycles × [denaturation at 94 °C for 30 s, primer annealing at 57 °C for 30 s, primer extension at 72 °C for 1 min] and a final extension step at 72 °C for 5 min. The products were electrophoresed on a 1% w/v agarose gel and the specific 740 bp band was gel extracted and purified using the Nucleospin PCR & Gel Clean-Up Kit (Macherey Nagel, Dueren, Germany) and sequenced using the Sanger method (CeMIA S.A.) with primer aegSCR8. Sequences were analysed using the sequence alignment editor BioEdit 7.2.5.
Analysis of the CHS-1 1043 locus
Analysis of the CHS-1 I1043 locus, to identify possible conserved DFB resistance mutations found in other species [29, 31], was performed in pools of mixed gDNA extracted individually from 5–8 Ae. albopictus samples of the same locality. Available Ae. albopictus DNA samples from other countries [28] were included in the analysis and genotyped individually. A 350-bp fragment of the CHS-1 gene, spanning the 1043 locus (numbering based on Musca domestica genomic sequence) was amplified in a 25 µl PCR (KAPA Taq PCR Kit) containing 1.5 µl DNA, 2.5 µl of 10× DNA polymerase buffer, 0.4 µM of each primer (primers kkv F3 and kkv R3; Additional file 1: Table S1), 0.4 µM of dNTPs and 1.5 U of Taq polymerase. The thermal conditions were: initial denaturation at 95 °C for 5 min, 40 cycles × [denaturation at 94 °C for 30 s, primer annealing at 55 °C for 30 s, primer extension at 72 °C for 1 min] and final extension step at 72 °C for 10 min. A small amount of the PCR products was electrophoresed on a 1.5% w/v agarose gel containing ethidium bromide to verify amplification. The remaining amount of the PCR products was purified using the Nucleospin PCR & Gel Clean-Up Kit (Macherey Nagel) and sequenced using the Sanger method (CeMIA S.A.) with the kkv F3 primer. Sequences were analysed using the sequence alignment editor BioEdit 7.2.5.
Metabolic resistance: detection of esterase gene amplification
CCEae3a and CCEae6a gene copy numbers were determined using quantitative real-time PCR on individual Ae. albopictus specimens. Amplification reactions at a 10 µl final volume were performed on a StepOnePlus Real-Time PCR System (Applied Biosystems, California, USA) containing 0.5 µl of gDNA, 0.2 µM of each primer (CCEae3aF, CCEae3aR, CCEae6aR and CCEEae6aF; Grigoraki et al. [28]; Additional file 1: Table S1) and SYBR Select Master Mix (Applied Biosystems, ThermoFisher Scientific, California, USA). Histone 3 (GenBank: XM_019687528.2) was used as a reference gene for normalization (primers His3 TaqF and His3 TaqR; Additional file 1: Table S1). The thermal parameters were: 50 °C for 2 min; 95 °C for 2 min, and 40 cycles × [95 °C for 3 s, 60 °C for 30 s]. Melting curves were performed for reference and target genes to verify the presence of a unique specific PCR product, which was checked on a 1% w/v agarose gel. A no-template control was included to detect possible contamination. Two replicates per sample were included. CCEae3a and CCEae6a gene copy numbers were estimated relatively to a temephos susceptible Αe. albopictus laboratory strain from Greece.
{kind=link}