Necropsy, bacteria isolation and identification
General fibrinous serositis was observed in dead birds from flocks A and B. In case of flock C, fibronecrotic typhlitis was the main gross pathological lesion found, a similar observation as in the 13-day old broiler from Portugal [7]. Omphalitis was diagnosed in turkey poults from flock D, while the chicken from a layer parent flock E did lack signs of bacterial infections but had a fracture of the leg. Escherichia coli was isolated from all cases investigated as reported by [7] who detected the same bacterium with M. morganii in liver and spleen of a broiler. Riemerella anatipestifer was detected in some organ samples from birds of flock B and Clostridium perfringens was found in association with a fibronecrotic typhlitis. When investigating the E. coli isolates for antibiotic resistance by disc diffusion method, smaller colonies were observed within the inhibition zone of ampicillin, amoxicillin and colistin, pointing towards a co-infection of E. coli and M. morganii. MALDI-TOF MS identified all strains to be M. morganii species, of which the strain PA17/10312 was classified as subspecies sibonii and the other four strains as subspecies morganii with a log score value above 2.5 (Table 1). On COS agar M. morganii could not be differentiated from E. coli or Salmonella spp., as both grow in round shape and appear as greyish smooth colonies. But on McConkey and Coliform agar, colonies were colourless and white, respectively, also very similar to Salmonella spp. Thus, morphological similarities to E. coli and Salmonella might have an impact on identification of M. morganii in routine diagnostics.
Antibiotic susceptibility testing
All isolates were found to be multidrug resistant (resistance to more than three antimicrobial substances) by disc diffusion method with resistance patterns ranging from five to ten antibiotics (Table 2). All were resistant to amoxicillin, ampicillin, and colistin, but also to tilmicosin and tylosin. The combination of trimethoprim/sulfamethoxazole was the only antibiotic to which all five strains were susceptible. Resistance to β-lactam antibiotics in Morganella species is usually mediated by the presence of chromosomally encoded β-lactamases belonging to the AmpC β-lactamase family [28]. These β-lactamases are typically induced in the presence of β-lactam antibiotics. The strain PA17/10312 (flock A) was confirmed to be an AMP-C phenotype. In contrast to ESBLs, AmpC hydrolyses broad and extended-spectrum cephalosporins (cephamycins as well as to oxyimino-β-lactams) but are not inhibited by β-lactamase inhibitors such as clavulanic acid [29]. The strains PA18/15564 (flock B) and PA18/25921 (flock D) were confirmed to be ESBL phenotype and possible productors of type D carbapenemases or impermeability/porin loss, whereas the strains PA18/16407 (flock C) and strain PA19/9695 (flock E) were ESBL negative. Overall, the results are in agreement with earlier data describing ESBL producers as multiple drug resistant [30].
Sequencing and annotation of five poultry strains
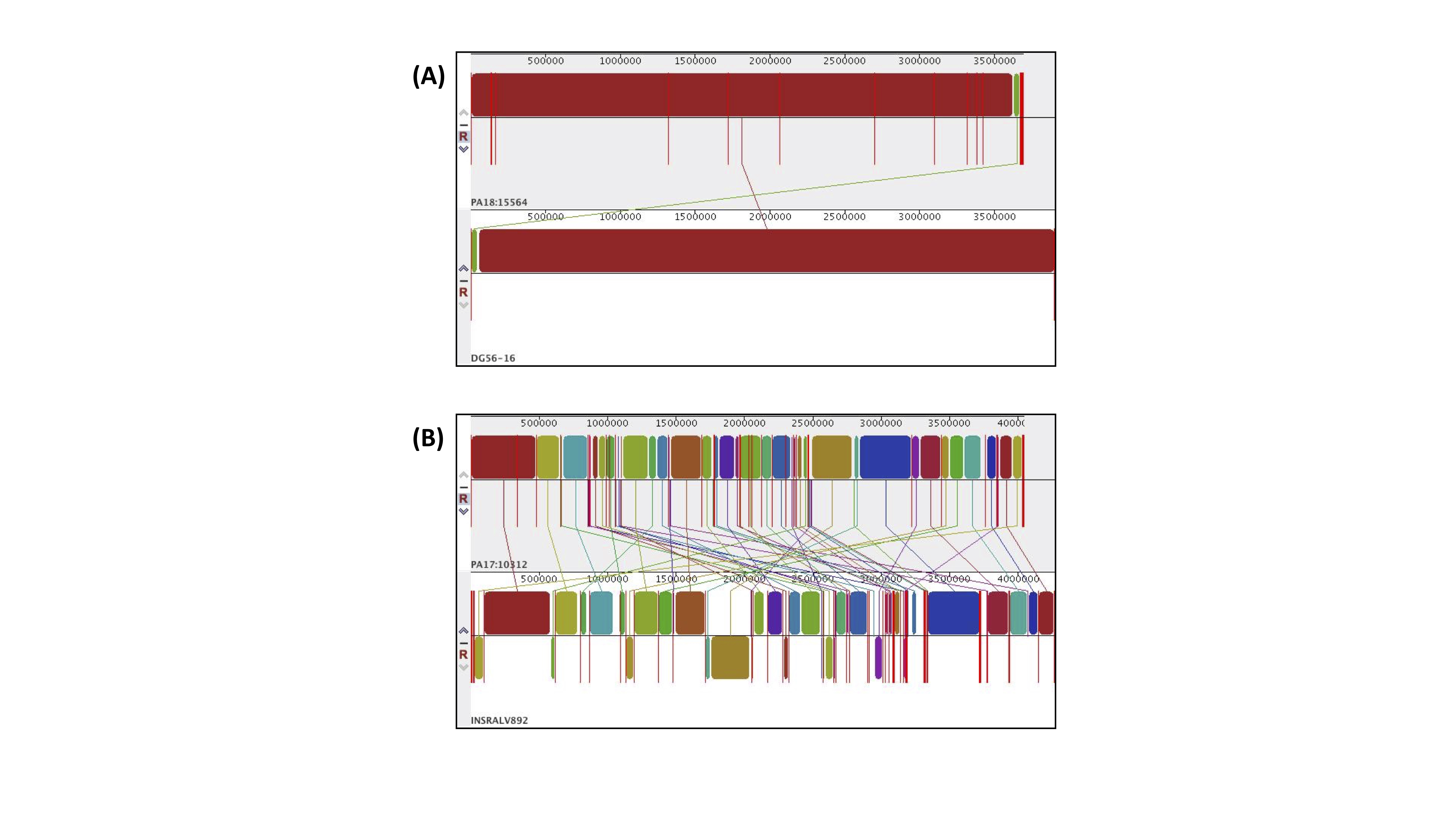
The genome sequences of the five poultry strains displayed substantial variation in length and number of genes (Table 3), with the PA17/10312 being longer and having more genes compared to the other strains. To evaluate whether these differences might be due to missing annotations, we computed genome completeness using the program CheckM [31] for each assembly and obtained values of 100% for all five strains, implying that the observed variation in gene content cannot be attributed to uneven coverage of the assembled strains. Genome size and gene content were also comparable to the assemblies of other published strains [7–10]. Additionally, we observed variations in genome structure among the five strains (Fig. 1A): PA18/15564 and PA18/16407 showed high colinearity compared to the closest reference strain (Additional File 2: Supplementary Fig. 1A), while PA17/10312 displayed more genomic rearrangements (Additional File 2: Supplementary Fig. 1B). Finally, the genome of the five strains was aligned to the reference strain KT, for which the most comprehensive annotation is available [8]. A multiple alignment in circular form is displayed in Fig. 1B and shows a high level of conservation among the five poultry strains, with few shared gaps, corresponding to the rRNA ribosomal gene clusters, which are notoriously difficult to assemble due to their repetitive nature.
Phylogenetic analysis
To test the hypothesis of a phylogenetic clustering based on the host in M. morganii, a phylogenetic tree was constructed including the five sequenced poultry isolates together with all available complete genomes from NCBI, encompassing a total of 52 strains. The genome-based k-mer tree displayed two distinct subtrees (M1 and M2), with no apparent clustering according to the host they were isolated from (Fig. 2). Lack of association with host was also previously reported for Salmonella enteritidis [12] and Providencia species [13]. The resulting tree places the strains PA18/16407, PA18/15564, PA19/9695, PA18/25921 in cluster M1 and the PA17/10312 strain in cluster M2, consistent with the differences in genome size and number of genes previously described. Historically, M. morganii has been divided into two subspecies: morganii and sibonii, based on the ability to ferment trehalose and other biochemical features [32]. Thus, we hypothesized that clusters M1 and M2 simply reflect the known subspecies classification. To test this idea, the presence of the trehalose operon (treR, treB and treP) in all the strains was investigated. By this approach, the subspecies sibonii was assigned to the strains in which the trehalose operon was present. Comparing this classification to the observed phylogenetic clustering, we found that the sibonii strains were entirely allocated in cluster M2, except the H1R strain (Fig. 2), confirming that differentiation of subspecies can be mostly performed based on phylogenetic grouping. These results are consistent with the subspecies assignment achieved by MALDI-TOF MS (Table 1). To validate the accuracy of the k-mer tree reconstruction, an additional tree was built using a whole-genome based approach (Additional File 3: Supplementary Fig. 2). Comparison of both trees showed high similarity in topology and few differences: i) the NCTC12358 and TW17014 strains of cluster M2 are swapped in the whole-genome based tree compared to the k-mer tree; ii) the 640_MMOR strain is grouped together with NCTC12286 and 8066 in the whole-genome based tree, while in the k-mer tree constitutes a separate cluster. As these differences do not affect the conclusions derived from following analyses, the k-mer tree was used as a guide for the remainder of the analyses.
Detection of resistance genes
The sequences of all five poultry strains contained genes for resistance to aminocoumarins, beta-lactams, elfamycins, fluoroquinolones, rifampin and tetracycline. Resistance to phenicol was present in all the sequenced poultry strains, except in the chicken strain PA19/9695. Based on BLAST searches against the MEGARes database, the investigation of presence/absence of resistance genes in all 52 strains from Fig. 2 revealed that resistance to certain antibiotics is widespread in Morganella. This includes aminocoumarins, beta-lactams, elfamycins, fluoroquinolones, phenicol, rifampin and tetracyclines (Fig. 3). Other types of resistance show a more uneven distribution along the tree, with multiple cases of strain-specific gains, like resistance to glycopeptides, which is present in 5/52 strains with no apparent phylogenetic clustering. While this might suggest a putative origin by horizontal gene transfer, a comprehensive analysis of mechanisms of originations of resistance genes is outside the scope of this study. Additionally, we looked at resistance genes that were absent in the MEGARes database, but that were previously reported for other strains of M. morganii, such as genes involved in lipid A biosynthesis (lpxA, lpxB, lpxC, lpxD, lpxH, lpxK, lpxL, lpxM, lpxP and lpxT), which are responsible for colistin resistance [9]. Such analyses detected lpxABCDHKLMPT in both geese strains, lpxABCDHKLMT in the chicken strain PA19/9695, lpxABCDHKLMT in the turkey strain PA18/25921 and lpxABCDHLMT in the goose strain PA17/10312. An additional copy of the lpxP gene was found in PA18/25921 and PA17/10312. Also, the tetB gene responsible for tetracycline resistance was found in all five poultry strains. No association was detected between resistance genes of a certain class and a specific phylogenetic cluster. These results are concordant with the phenotypic tests of antibiotic resistance (Table 2). Overall, the classes of resistance determined are consistent with a previously compiled review of resistance genes in M. morganii [3].
Detection of known virulence genes
In an initial approach, the sequences from the poultry strains were screened for known virulence genes based on the comparison with the KT strain, for which an exhaustive list of virulence genes is available. However, the gene-id based detection of genes from the KT strain was unfeasible due to incongruences between gene-ids from the KT strain and the ones deposited in the NCBI gene database, thus the 4-letter gene name was used to retrieve the genes of interest, resulting in a partial set of 125 genes for this analysis. Most of the known virulence genes present in this set were found in the sequences of the five poultry strains (Fig. 4). This not only indicates that virulence genes are very conserved, but also suggests that the annotations of the five poultry strains is highly complete. As this gene set did not include all known virulence genes, we additionally looked for the presence of specific classes of virulence genes of importance. Hemolysins are essential for pore-formation during invasion of host cells [33] and encoded by the hlyCABD operon. This operon was present and intact only in the chicken strain PA18/25921, while absent in PA18/16407, PA18/15564 and PA19/9695. In the sibonii strain PA17/10312, only hlyC and hlyD were present and distributed on different contigs. Variation in hemolysin composition was also observed in three recently sequenced human strains [10]. Ureases are also typically organized into the ureABCFGD operon and contribute to formation of urinary stones [34]. This operon was present and intact in all five strains. Finally, the capsule synthesis regulation genes rcsB, rcsC, rcsD and rcsF involved in host immune response [35] were also detected in all five strains, with an additional copy of rcsB in PA18/25921. Interestingly, virulence genes in the category “toxins” displayed high variation in copy numbers among the five strains (Fig. 4). Manual scanning of the alignments also showed substantial variation in coding sequences exemplarily shown for the RtxA toxin in Additional File 4: Supplementary Fig. 3. For these reasons, a positive selection scan using PAML (see Materials and methods) was performed based on the hypothesis that toxins might evolve under positive selection in M. morganii. For this analysis only toxins present in all five strains were selected (Fig. 4) and it was found that four out of nine toxins (tcaC/tcdB2, xptA1, xptB1, xptC1) showed signs of positive selection in the PA17/10312 branch (Table 4). Notably, the insecticidal proteins XptA1B1C1 had the strongest signal of positive selection. While the precise function of these toxins in M. morganii is unknown, it was shown in Xenorhabdus spp. and Photorhabdus luminescens that they facilitate the killing of the insect host via nematodes living in symbiosis with these bacteria [36]. Thus, positive selection in insecticidal toxins might reflect an evolutionary arms race among species.
Detection of novel virulence genes
To identify novel virulence genes, the proteomes of the five poultry strains were BLASTed to a curated set of Enterobacteriaceae virulence factors generated from the Virulence Factor DataBase (VFDB). For each strain, presence/absence of each virulence gene was computed, and virulence genes were classified according to different functional categories based on the VFDB functional annotation. To get an overview of the variation in individual virulence genes across the strains, the number of virulence genes of different functional categories in each strain was plotted sorted according to the phylogenetic position on the tree (Fig. 5). An increase in number of genes in strains of cluster M2 compared to the cluster M1 in the virulence categories adhesion, secretion system and hypothetical was observed (Fig. 5). As the strain PA17/10312 is also located in cluster M2, these results might explain the higher number of genes in the PA17/10312 strain compared to the other strains. Novel virulence genes were defined as those specific to cluster M2: a total of 39 novel virulence genes were found in this way (Table 4). To validate specificity to cluster M2, these genes were BLASTed to two outgroup species: Providencia stuartti and Proteus mirabilis, and only genes with no hits to both species were retained. This resulted in a final set of 26 novel virulence genes. Among these, afaAB [37], pixC [38], sfaG and fyuA [39] are known to be associated with adhesion and colonization of urinary trait in E. coli, while the intimin fdeC [40], the Shiga toxins stx2A/stxA [41], escN [42], espD [43] are connected with enteropathogenic strains of E. coli. This might suggest that some M. morganii strains of the sibonii subspecies might have acquired the ability to invade the enteric tract. With regard to phylogenetic distribution, 10/26 genes were shared by all strains in cluster M2, consistent with ancestral gain in cluster M2 and 12/26 genes were specific to a single strain, likely due to strain-specific horizontal gene transfer events. To find evidence for horizontal gene transfer the novel virulence genes were BLASTed against the whole bacterial protein database. Then, using the program Alienness [44], evidence for horizontal gene transfer (HGT) was detected in 6/26 (23%) of the novel virulence genes (afaB, escU, escV, SG1030, stx2A, ycbV), having an Alien Index (AI) > 30 (Additional File 5: Supplementary Table 2). These genes were assigned to the different donors of the class Morganellaceae (Additional File 5: Supplementary Table 2). While horizontal gene transfer of resistance genes was previously described in M. morganii [9], to our knowledge this is the first report of HGT of virulence genes in M. morganii.
Prediction of pathogenic potential for humans
Using PathogenFinder 1.1 [45] the pathogenic potential of the five poultry strains for humans was estimated. The approach used by this tool is based on the presence of group of genes that are frequently associated with human pathogenic bacteria. Results showed that the PA17/10312 was predicted to be pathogenic in humans (Probability = 0.64), while the four other strains were classified as non-pathogenic in humans (Probability = 0.57 for PA18/15564, 0.52 for PA18/16407, 0.57 for PA18/25921 and 0.54 for PA19/9695). These results are consistent with the higher number of virulence factors found in PA17/10312 compared to the other strains. It remains to be determined in an animal trial whether these findings also reflects the host-specific pathogenicity.
Annotation of mobile elements
Mobile elements such as plasmids, prophages and integrons are often responsible for transfer of resistance genes in bacteria [46, 47]. No evidence for plasmids was found in any of the five poultry strains using two independent approaches (see Materials and methods). In the published strains, no plasmids were found in the KT strain [8] but were detected in the INSRALV892 strain [7], while no information is available for the other sequenced strains [9, 10]. A total of the four active prophages were detected in PA17/10312, three in PA18/15564, two in PA18/16407, five in PA18/25921 and one in PA19/9695 (Additional File 6: Supplementary Table 3), none of them showing association with resistance genes. Two integrons were found in the PA19/9695 strain and one integron was found in the PA17/10312 strain, also not associated with resistance genes. Pathogenicity islands are another class of mobile elements, which are linked with virulence genes in a large number of bacteria [48]. A total of 18 pathogenicity islands were detected in PA17/10312, 16 in PA18/15564, 21 in PA18/16407, 22 in PA18/25921 and 23 in PA19/9695 (Additional File 7: Supplementary Table 4), associated, respectively, with 25, 9, 23, 16 and 24 virulence genes. The most abundant virulence categories associated with these genes were: “adherence” for 20/25 genes in PA17/10312, “adherence” for 5/9 genes in PA18/15564 and “iron uptake” for 11/23 genes in PA18/16407, “toxins” for 5/16 genes in PA18/25921 and “adherence” for 11/24 in PA19/9695. In summary, these results suggest high variability in gene content of pathogenicity islands, probably due to phenomena of genome rearrangements and lateral transfer.
Identification of strain-specific metabolic pathways
To study metabolic pathways, the annotations of the five poultry strains were submitted to BlastKOALA providing a global overview of metabolic pathways displayed as a metabolic network. This program also allows an easy comparison of metabolic networks among different organisms or strains. All metabolic pathways were entirely conserved among the five poultry strains. In addition, two novel pathways specific to the PA17/10312 strain were discovered (Fig. 6): sucrose catabolism and biosynthesis of betaine from choline. Betaine is an osmoprotectant that allows organisms to grow in high-osmolarity environments [49]. Some bacteria can synthesize betaine de novo from intermediates of central metabolism whereas others can synthesize it only from exogenously supplied choline [50]. Screening all the 52 M. morganii strains included in Fig. 2 revealed that only the FAM24091 strain (isolated from Switzerland cheese), which is the closest strain to PA17/10312, possess genes for betaine biosynthesis (BetA and BetB). No evidence for horizontal gene transfer was found, based on the absence of proteins from closely related species which would cluster together with BetA and BetB from PA17/10312 based upon BLAST analyses. Whether the determined pathways are beneficial for the bacteria in vitro and in vivo remains speculative for the moment and needs to be determined in additional studies.
{kind=link}
{kind=link}
{kind=link}