Material and equipment
The synthesis of 1-(4-acetylphenyl)-3-alkylimidazolium salts (2a-g, 3a-f) was carried out in flame-dried glassware under an inert atmosphere using standard Schlenk techniques. Acetylcholinesterase from the electric eel, 5,5’-dithiobis-(2-nitrobenzoic) acid, acetylthiocholine iodide and tacrine were purchased from Sigma Aldrich. The solvents commercially purchased were used without exposure to any purification and drying process. All other reagents were commercially available by Merck, Alfa Aesar, Carlo Erba, and Sigma-Aldrich Chemical Co. and used without further purification. Melting points were identified in glass capillaries under air with an Electrothermal-9200 melting point apparatus. FT-IR spectra were saved in the range 400–4000 cm− 1 on Perkin Elmer Spectrum 100 FT-IR spectrometer. Carbon (13C), Proton (1H), and Fluorine (19F) NMR spectra were recorded using either a Bruker Avance III 400 MHz NMR spectrometer operating at NMR (400 MHz for 1H, 100 MHz for 13C NMR, and 376 MHz for 19F NMR, respectively) in the CDCl3 with tetramethylsilane (TMS) as an internal reference. All spectroscopic data in this study were performed by Inonu University Scientific and Technology Centre (Malatya, TÜRKİYE).
Synthesis
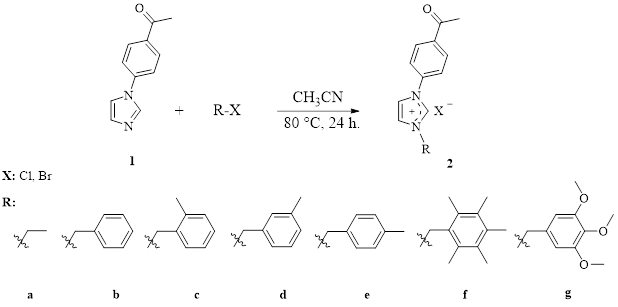
Synthesis of 1-(4-acetylphenyl)-3-ethylimidazolium bromide (2a)
The method described in the literature was used to synthesize compound 2a. 1-(4-acetylphenyl)imidazole (745 mg, 4 mmol) and ethyl bromide (436 mg, 4 mmol) were dissolved in acetonitrile (4 mL) and stirred for 24 hours at 80°C. The white solid that formed at the end of the reaction was filtered off, the solvent was removed, and the product was washed with diethyl ether. The crude product was crystallized from a mixture of ethyl alcohol/diethyl ether (1:3) [80]. Yield: 67% (791 mg). m.p.: 220–221°C; ν(C=O): 1678 cm− 1; ν(CN): 1551 cm− 1. Anal. calc. for C13H15BrN2O: C: 52.90; H: 5.12; N: 9.49. Found: C: 52.91; H: 5.14; N: 9.51. 1H NMR (400 MHz, CDCl3, 298 K), δ: 1.63 (t, 3H, J: 7.3 Hz, -NCH2CH3); 2.57 (s, 3H, -NC6H4COCH3); 4.58 (quar., 2H, J: 7.3 Hz, -NCH2CH3); 7.76 and 7.98 (s, 2H, imidazol-4,5-CH); 8.00 and 8.07 (d, 4H, J: 8.8 and 8.7 Hz, -NC6H4COCH3); 11.12 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 15.8 and 46.0 (-NCH2CH3); 26.9 (-C6H4COCH3); 120.7 ve 123.1 (imidazol-4,5-CH); 122.0, 130.6, 136.1 and 137.5 (Ar-C); 138.0 (NCHN); 196.5 (-NC6H4COCH3).
Synthesis Of 1-(4-acetylphenyl)-3-benzylimidazolium Chloride (2b)
The compound 2b was synthesized by using the same way for 2a. But benzyl chloride (506 mg, 4 mmol) was used instead of ethyl bromide. Yield: 79% (988 mg). m.p.: 184–185°C; ν(C=O): 1682 cm− 1; ν(CN): 1552 cm− 1. Anal. calc. for C18H17ClN2O: C: 69.12; H: 5.48; N: 8.96. Found: C: 69.10; H: 5.47; N: 8.97. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.51 (s, 3H, -C6H4COCH3); 5.71 (s, 2H, -NCH2C6H5); 7.65 and 8.06 (s, 2H, imidazol-4,5-CH); 7.27 (m, 3H, -NCH2C6H5); 7.56 (m 2H, -NCH2C6H5); 7.72–7.79 (m, 1H, -NC5H5); 7.89–8.03 (m, 4H, -NC6H4COCH3); 11.64 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 26.8 (-NC6H4COCH3); 53.6 (-NCH2C6H5); 120.7 and 123.1 (imidazol-4,5-CH); 121.6, 123.1, 129.3, 129.4, 129.6, 130.6, 133.0, 136.6 and 137.6 (Ar-C); 137.8 (NCHN); 196.4 (-NC6H4COCH3).
Synthesis Of 1-(4-acetylphenyl)-3-(2-methylbenzyl)imidazolium Chloride (2c)
The compound 2c was synthesized by using the same way for 2a. But 2-methylbenzyl chloride (562 mg, 4 mmol) was used instead of ethyl bromide. Yield: 84% (1.10 g). m.p.: 205–206°C; ν(C=O): 1665 cm− 1; ν(CN): 1545 cm− 1. Anal. calc. for C19H19ClN2O: C: 69.83; H: 5.86; N: 8.57. Found: C: 69.81; H: 5.88; N: 8.56. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.32 (s, 3H, -NCH2C6H4CH3-2); 2.55 (s, 3H, -NC6H4COCH3); 5.74 (s, 2H, -NCH2C6H4CH3-2); 7.16–7.20 (m, 4H, -NCH2C6H4CH3-2); 7.25 and 7.39 (d, 2H, J: 7.5 and 7.2 Hz, imidazol-4,5-CH); 7.97 and 8.05 (d, 4H, J: 8.3 and 8.1 Hz, -NC6H4COCH3); 11.74 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 19.4 (-NCH2C6H4CH3-2); 26.8 (-NC6H4COCH3); 52.1 (-NCH2C6H4CH3-2); 120.3 and 122.4 (imidazol-4,5-CH); 121.8, 122.2, 127.1, 130.1, 130.4, 130.6, 130.7, 131.4, 137.4 and 137.6 (Ar-C); 138.0 (NCHN); 196.3 (-NC6H4COCH3).
Synthesis Of 1-(4-acetylphenyl)-3-(3-methylbenzyl)imidazolium Chloride (2d)
The compound 2d was synthesized by using the same way for 2a. But 3-methylbenzyl chloride (562 mg, 4 mmol) was used instead of ethyl bromide. Yield: 79% (1.03 g). m.p.: 174–175°C; ν(C=O): 1682 cm− 1; ν(CN): 1552 cm− 1. Anal. calc. for C19H19ClN2O: C: 69.83; H: 5.86; N: 8.57. Found: C: 69.82; H: 5.87; N: 8.58. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.26 (s, 3H, -NCH2C6H4CH3-3); 2.54 (s, 3H, -C6H4COCH3); 5.67 (s, 2H, -NCH2C6H4CH3-3); 7.12 (d, 2H, J: 7.1 Hz, -NCH2C6H4CH3-3); 7.31 (s, 2H, -NCH2C6H4CH3-3); 7.45 (s, 1H, imidazol-4,5-CH); 7.85 (d, 1H, J: 8.8 Hz, imidazol-4,5-CH); 7.95 (d, 2H, J: 8.4 Hz, -NC6H4COCH3); 8.02 (s, 2H, -NC6H4COCH3); 11.76 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 21.3 (-NCH2C6H4CH3-3); 26.8 (-NC6H4COCH3); 53.9 (-NCH2C6H4CH3-3); 120.3 and 122.7 (imidazol-4,5-CH); 121.7, 126.4, 129.4, 129.9, 130.5, 130.7, 132.6, 137.1, 137.5 and 138.0 (Ar-C); 139.5 (NCHN); 196.3 (-NC6H4COCH3).
Synthesis Of 1-(4-acetylphenyl)-3-(4-methylbenzyl)imidazolium Chloride (2e)
The compound 2e was synthesized by using the same way for 2a. But 4-methylbenzyl chloride (562 mg, 4 mmol) was used instead of ethyl bromide. Yield: 78% (1.02 g). m.p.: 187–188°C; ν(C=O): 1665 cm− 1; ν(CN): 1545 cm− 1. Anal. calc. for C19H19ClN2O: C: 69.83; H: 5.86; N: 8.57. Found: C: 69.84; H: 5.84; N: 8.57. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.26 (s, 3H, -NCH2C6H4CH3-4); 2.53 (s, 3H, -NC6H4COCH3); 5.66 (s, 2H, -NCH2C6H4CH3-4); 7.11 and 7.42 (d, 4H, J: 6.7 and 7.5 Hz, -NCH2C6H4CH3-4); 7.48 and 7.88 (s, 2H, imidazol-4,5-CH); 7.94 and 8.02 (d, 4H, J: 8.1 and 7.5 Hz, -NC6H4COCH3); 11.72 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 21.1 (-NCH2C6H4CH3-4); 26.7 (-NC6H4COCH3); 53.6 (-NCH2C6H4CH3-4); 120.3 and 122.7 (imidazol-4,5-CH); 121.7, 129.4, 129.8, 130.2, 130.6, 136.9, 137.6 and 137.9 (Ar-C); 139.8 (NCHN); 196.3 (-NC6H4COCH3).
Synthesis Of 1-(4-acetylphenyl)-3-(2,3,4,5,6-pentamethylbenzyl)imidazolium Chloride (2f)
The compound 2f was synthesized by using the same way for 2a. But 2,3,4,5,6-pentamethylbenzyl chloride (787 mg, 4 mmol) was used instead of ethyl bromide. Yield: 81% (1.24 g). m.p.: 242–243°C; ν(C=O): 1683 cm− 1; ν(CN): 1555 cm− 1. Anal. calc. for C23H27ClN2O: C: 72.14; H: 7.11; N: 7.32. Found: C: 72.12; H: 7.10; N: 7.30. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.17 and 2.21 (s, 15H, -NCH2C6(CH3)5-2,3,4,5,6); 2.54 (s, 3H, -NC6H4COCH3); 5.81 (s, 2H, -NCH2C6(CH3)5-2,3,4,5,6); 6.86 and 7.82 (s, 2H, imidazol-4,5-CH); 8.00 and 8.07 (d, 4H, J: 8.4 and 8.2 Hz, -NC6H4COCH3); 11.84 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 16.9 and 17.3 (-NCH2C6(CH3)5-2,3,4,5,6); 26.4 (-C6H4COCH3); 49.9 (-NCH2C6(CH3)5-2,3,4,5,6); 121.6 and 125.0 (imidazol-4,5-CH); 119.8, 121.8, 130.7, 133.8, 133.9, 137.5, 137.7 and 137.4 (Ar-C); 138.0 (NCHN); 196.3 (-NC6H4COCH3).
Synthesis Of 1-(4-acetylphenyl)-3-(3,4,5-trimethoxybenzyl)imidazolium Chloride (2g)
The compound 2g was synthesized by using the same way for 2a. But 3,4,5-trimethoxybenzyl chloride (864 mg, 4 mmol) was used instead of ethyl bromide. Yield: 75% (1.21 g). m.p.: 222–223°C; ν(C=O): 1681 cm− 1; ν(CN): 1555 cm− 1. Anal. calc. for C21H23ClN2O4: C: 62.61; H: 5.75; N: 6.95. Found: C: 62.59; H: 5.74; N: 6.93. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.54 (s, 3H, -NC6H4COCH3); 3.75 and 3.82 (s, 9H, -NCH2C6H2(OCH3)3-3,4,5); 5.62 (s, 2H, -NCH2C6H4(OCH3)3-3,4,5); 6.93 (s, 2H, NCH2C6H2(OCH3)3-3,4,5); 7.74 and 7.60 (s, 2H, imidazol-4,5-CH); 7.91 and 8.02 (d, 4H, J: 8.6 and 8.4 Hz, -NC6H4COCH3); 11.73 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 26.8 (-NC6H4COCH3); 54.1 (-NCH2C6H4(OCH3)3-3,4,5); 56.7 and 60.8 (-NCH2C6H4(OCH3)3-3,4,5); 121.3 and 122.9 (imidazol-4,5-CH); 107.0, 119.9, 121.7, 128.3, 130.5, 130.6, 137.0, 137.5, 138.0 and 153.9 (Ar-C); 138.9 (NCHN); 196.3 (-NC6H4COCH3).
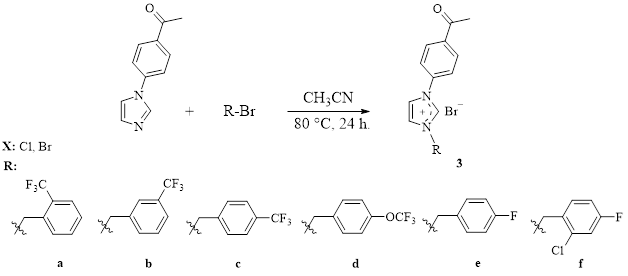
Synthesis Of 1-(4-acetylphenyl)-3-(2-trifluoromethylbenzyl)imidazolium Bromide (3a)
The compound 3a was synthesized by using the same way for 2a. But 2-trifluoromethylbenzyl bromide (717 mg, 3 mmol) was used instead of ethyl bromide. Yield: 77% (982 mg). m.p.: 234–235°C; ν(C=O): 1678 cm− 1; ν(CN): 1554 cm− 1. Anal. calc. for C19H16BrF3N2O: C: 53.66; H: 3.79; N: 6.59. Found: C: 53.67; H: 3.80; N: 6.58. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.55 (s, 3H, -NC6H4COCH3); 5.92 (s, 2H, -NCH2C6H4CF3-2); 7.27 (s, 1H, -NCH2C6H4CF3-4); 7.49 (t, 1H, J: 7.7 Hz, -NCH2C6H4CF3-2); 7.61 (t, 1H, J: 7.6 Hz, -NCH2C6H4CF3-2); 7.80 (s, 1H, -NCH2C6H4CF3-2); 7.68 and 8.13 (d, 2H, J: 7.8 and 7.7 Hz, imidazol-4,5-CH); 7.97 and 8.06 (d, 4H, J: 8.6 and 8.6 Hz, -NC6H4COCH3); 11.34 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 26.8 (-NC6H4COCH3); 50.1 (-NCH2C6H4CF3-2); 120.5 and 122.8 (imidazol-4,5-CH); 122.1, 125.4, 126.6, 128.7, 129.0, 130.2, 130.3, 130.7, 133.4, 133.6, 136.9 and 137.4 (Ar-C and CF3); 138.2 (NCHN); 196.2 (-NC6H4COCH3). 19F NMR (376 MHz, CDCl3, 298 K), δ (ppm): −58.38 (Ar–CF3).
Synthesis Of 1-(4-acetylphenyl)-3-(3-trifluoromethylbenzyl)imidazolium Bromide (3b)
The compound 3b was synthesized by using the same way for 2a. But 3-trifluoromethylbenzyl bromide (717 mg, 3 mmol) was used instead of ethyl bromide. Yield: 80% (1.02 g). m.p.: 204–205°C; ν(C=O): 1676 cm− 1; ν(CN): 1551 cm− 1. Anal. calc. for C19H16BrF3N2O: C: 53.66; H: 3.79; N: 6.59. Found: C: 53.68; H: 3.78; N: 6.58. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.57 (s, 3H, -NC6H4COCH3); 5.91 (s, 2H, -NCH2C6H4CF3-3); 7.43 and 7.62 (s, 2H, -NCH2C6H4CF3-3); 7.53 and 7.61 (d, 1H, J: 7.7 Hz, -NCH2C6H4CF3-3); 7.73 (s, 1H, imidazol-4,5-CH); 7.96 (d, 1H, J: 7.3 Hz, imidazol-4,5-CH); 7.90 and 8.09 (d, 4H, J: 8.4 and 8.2 Hz, -NC6H4COCH3); 11.56 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 26.8 (-NC6H4COCH3); 53.0 (-NCH2C6H4CF3-3); 120.3 and 122.7 (imidazol-4,5-CH); 111.8, 122.0, 125.8, 126.2, 130.5, 130.8, 133.7 and 137.2 (Ar-C and CF3); 138.4 (NCHN); 196.3 (-NC6H4COCH3). 19F NMR (376 MHz, CDCl3, 298 K), δ (ppm): −60.97 (Ar–CF3).
Synthesis Of 1-(4-acetylphenyl)-3-(4-trifluoromethylbenzyl)imidazolium Bromide (3c)
The compound 3c was synthesized by using the same way for 2a. But 4-trifluoromethylbenzyl bromide (717 mg, 3 mmol) was used instead of ethyl bromide. Yield: 78% (995 mg). m.p.: 195–196°C; ν(C=O): 1678 cm− 1; ν(CN): 1551 cm− 1. Anal. calc. for C19H16BrF3N2O: C: 53.66; H: 3.79; N: 6.59. Found: C: 53.65; H: 3.77; N: 6.60. 1H NMR (400 MHz, CDCl3, 298 K), δ: 1H NMR (400 MHz, DMSO), δ: 2.55 (s, 3H, -NC6H4COCH3); 5.92 (s, 2H, -NCH2C6H4CF3-4); 7.57 and 7.78 (d, 4H, J: 8.0 and 8.2 Hz, -NCH2C6H4CF3-4); 7.58 and 7.69 (s, 2H, imidazol-4,5-CH); 7.88 and 8.04 (d, 4H, J: 8.7 and 8.6 Hz, -NC6H4COCH3); 11.35 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 26.8 (-NC6H4COCH3); 53.0 (-NCH2C6H4CF3-4); 120.4 and 123.1 (imidazol-4,5-CH); 122.0, 126.4, 126.5, 130.0, 130.7, 136.7 and 137.4 (Ar-C and CF3); 138.3 (NCHN); 196.2 (-NC6H4COCH3). 19F NMR (376 MHz, CDCl3, 298 K), δ (ppm): −61.10 (Ar–CF3).
Synthesis Of 1-(4-acetylphenyl)-3-(4-trifluoromethoxybenzyl)imidazolium Bromide (3d)
The compound 3d was synthesized by using the same way for 2a. But 4-trifluoromethoxybenzyl bromide (765 mg, 3 mmol) was used instead of ethyl bromide. Yield: 73% (966 mg). m.p.: 194–195°C; ν(C=O): 1682 cm− 1; ν(CN): 1552 cm− 1. Anal. calc. for C19H16BrF3N2O2: C: 51.72; H: 3.66; N: 6.35. Found: C: 531.70; H: 3.67; N: 6.33. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.53 (s, 3H, -NC6H4COCH3); 5.83 (s, 2H, -NCH2C6H4OCF3-4); 7.14 and 7.73 (d, 4H, J: 8.1 and 8.6 Hz, -NCH2C6H4OCF3-4); 7.71 and 7.80 (s, 2H, imidazol-4,5-CH); 7.89 and 8.02 (d, 4H, J: 8.7 and 8.6 Hz, -C6H4COCH3); 11.23 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 26.7 (-NC6H4COCH3); 52.7 (-NCH2C6H4OCF3-4); 120.6 and 123.3 (imidazol-4,5-CH); 119.0, 121.7, 121.9, 130.6, 131.4, 131.6, 136.2, 137.4 and 150.1 (Ar-C and OCF3); 138.2 (NCHN); 196.2 (-C6H4COCH3). 19F NMR (376 MHz, CDCl3, 298 K), δ (ppm): −56.79 (Ar–OCF3).
Synthesis Of 1-(4-acetylphenyl)-3-(4-fluorobenzyl)imidazolium Bromide (3e)
The compound 3e was synthesized by using the same way for 2a. But 4-fluorobenzyl bromide (752 mg, 4 mmol) was used instead of ethyl bromide. Yield: 82% (1.23 g). m.p.: 167–168°C; ν(C=O): 1672 cm− 1; ν(CN): 1552 cm− 1. Anal. calc. for C18H16BrFN2O: C: 57.62; H: 4.30; N: 7.47. Found: C: 57.60; H: 4.31; N: 7.46. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.54 (s, 3H, -NC6H4COCH3); 5.75 (s, 2H, -NCH2C6H4F-4); 6.98 (t, 2H, J: 8.5 Hz, -NCH2C6H4F-4); 7.63–7.65 (m, 2H, -NCH2C6H4F-4); 7.67 and 7.83 (s, 2H, imidazol-4,5-CH); 7.90 and 8.02 (d, 4H, J: 8.6 and 9.0 Hz, -NC6H4COCH3); 11.66 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 26.7 (-NC6H4COCH3); 52.9 (-NCH2C6H4F-4); 120.4 and 122.9 (imidazol-4,5-CH); 116.4, 116.6, 121.1, 121.7, 129.0, 130.4, 130.6, 131.8, 136.8, 137.5 and 137.4 (Ar-C); 138.0 (NCHN); 162.1 and 164.6 (Ar-C-F); 196.3 (-NC6H4COCH3). 19F NMR (376 MHz, CDCl3, 298 K), δ (ppm): −112.96 (Ar–F).
Synthesis Of 1-(4-acetylphenyl)-3-(2-chloro-4-fluorobenzyl)imidazolium Bromide (3f)
The compound 3f was synthesized by using the same way for 2a. But 2-chloro-4-fluorobenzyl bromide (670 mg, 3 mmol) was used instead of ethyl bromide. Yield: 74% (910 mg). m.p.: 168–169°C; ν(C=O): 1679 cm− 1; ν(CN): 1554 cm− 1. Anal. calc. for C19H15BrFClN2O: C: 52.77; H: 3.69; N: 6.84. Found: C: 52.80; H: 3.70; N: 6.86. 1H NMR (400 MHz, CDCl3, 298 K), δ: 2.56 (s, 3H, -NC6H4COCH3); 5.88 (s, 2H, -NCH2C6H3ClF); 7.03 (t, 1H, J: 7.0 Hz, -NCH2C6H3ClF); 7.11 (d, 1H, J: 2.0 Hz, -NCH2C6H3ClF); 7.50 (d, 1H, J: 8.5 Hz, -NCH2C6H3ClF); 7.54 and 7.77 (s, 2H, imidazol-4,5-CH); 7.92 and 8.06 (d, 4H, J: 8.4 and 8.3 Hz, -NC6H4COCH3); 11.66 (s, 1H, NCHN). 13C NMR (100 MHz, CDCl3, 298 K), δ: 26.8 (-NC6H4COCH3); 50.3 (-NCH2C6H4ClF); 120.0 and 122.9 (imidazol-4,5-CH); 115.9, 117.4, 120.9, 121.8, 127.0, 130.4, 130.7, 134.9 and 137.6 (Ar-C); 138.1 (NCHN); 162.0 and 164.6 (Ar-C-F); 196.2 (-NC6H4COCH3). 19F NMR (376 MHz, CDCl3, 298 K), δ (ppm): −110.06 (Ar–F).
Biochemical Studies
AChE inhibition assay
In the present work, in vitro effects of acetylphenyl-substituted imidazolium salts (2a-g, 3a-f) and 1-(4-acetylphenyl)imidazole (1) on AChE activity evaluated by Ellman method [81] as described in prior studies [82, 83]. Results were performed spectrophotometrically at 412 nm using acetylthiocholine iodide according to previous study [84].
Hcas Inhibition Assay
Commercial sources usually isolate both CA isoenzymes from fresh human erythrocytes and typically produce these isoenzymes by recombinant technology from E. coli [85]. In this study, both hCA isoenzymes were purified via Sepharose-4B-L-Tyrosine-sulfanilamide affinity chromatography [86]. CA isoenzymes were purified by Sepharose-4B-L-Tirozyne-sulfanylamide affinity column chromatography [87]. The protein content during the purification steps was determined by Bradford method [88]. Also, bovine serum albumin was used as standard protein [89]. The purity of both hCA isoenzymes was controlled by SDS-PAGE as described in prior studies [90]. During the isoenzyme’s purification and inhibition process, esterase activity was performed [91]. Both hCA isoenzyme activities were determined by following the change in absorbance at 348 nm according to the assay by Verpoorte et al. [92] as described in detail [93].
Ache And Hcas Kinetic Assay
To investigate the in vitro inhibitory mechanisms of acetylphenyl-substituted imidazolium salts (2a-g, 3a-f) and 4-(1-H-imidazol-1-yl)acetophenone (1) kinetic studies were made with the variable compound and substrate concentrations [94, 95]. From the observed data, IC50 and Ki values for these derivatives were computed, and the types of inhibition of AChE and hCAs were determined as in previous studies [96].
In Silico Studies
The 3D structures of carbonic anhydrases (hCA-I, hCA-II) and acetylcholinesterase (AChE) were obtained from the RCSB Protein Data Bank (PDB) having identifiers 1AZM (2.00 Å), 3HS4 (1.10 Å), and 4EY6 (2.40 Å), respectively. The 3D structures of proteins were evaluated in Discovery Studio 3.5 [97], and missing residue(s) and polar hydrogen atoms were added, and optimized by CHARMM force field [98] of DS 3.5 software. The data on ligand-binding sites in all three enzyme proteins were obtained from the literatures and with help of the “define and edit binding site” sub-protocol of DS software. Also the potential first three synthesized imidazolium salts as ligands were sketched and minimized at DFT/B3LYP/SDD level in Gaussian 09 (G09) [99]. Lastly, the current ligands were docked into the related ligand-binding sites of each target using AutoDock 4.2 software [100]. Intermolecular interactions among enzymes and ligands were assessed and 3D images were rendered with help of DS 3.5 software.
As control compounds, molecular docking calculations were also applied the drugs acetazolamide (AZA) and tacrine (TAC) for among carbonic anhydrases (hCA-I, hCA-II) and acetylcholinesterase (AChE), respectively. The data of the control compounds were also utilized to compare and analyze the selected ligands.
Besides these, the pharmacokinetics features of docking-based prioritized molecules as ligands ADMET were examined to define their activity within the human body via were calculated using DS 3.5 [97]. The Lipinski [101] and Veber Rules [102] were used to estimate the drug-like qualities of the ligands. These rules are generally used and important rules in the pharmaceutical industry as a first step in describing the optimal compound for a medicine. So that, the bioavailability of the compounds can be forecasted [103]. After then, ADMET descriptors [Aqueous solubility, intestinal absorption (HIA), blood brain barrier penetration (BBB), and cytochrome P450 2D6 binding (CYP2D6), hepatotoxicity and plasma protein binding (PPB)] were predicted with sub-protocol of DS 3.5 to be suitable drug properties or not of the current compounds.
{kind=link}
{kind=link}