We propose the cooperation between autophagy and LD that positively contributes to ZIKV replication. Since the ZIKV genome does not have the machinery necessary for lipid synthesis, these lipids are derived from the host cell ER membrane. In our study, infected cells had the most LD seen at 12 hpi and the lowest at 48 hpi (Fig. 1B). Viral transcription was highest at 48 hpi (Fig. 1C). This dependence on lipids for flavivirus life cycle has been documented for several viruses (25–27). We also report, similarly to Chen et al (28), an increase in the amount and size of the LD in uninfected neighboring cells compared to infected ones (Supplemental Fig. 1S). This phenomenon is shown by several viruses, interpreted by others as indicating either the establishment of intercellular channels within the gap junctions or soluble mediators secreted or taken up by infected cells (29–31). Thus, ZIKV infection not only directly regulates the lipid metabolism to support the virus life cycle but also induces bystander effects. Variations in LD distribution, viral transcription level changes with time, and the bystander effect all suggest that ZIKV uses host lipids to form its replication complexes and alter the metabolism of surrounding cells to propagate its infection.
LD availability can determine the amount of viral infection. Treatment with ATV reduces the amount of LD (Fig. 2A) and impairs viral transcription, translation, and release (Fig. 2B-D). Statins and their derivatives disrupt infection by ZIKV and other flaviviruses (32, 33). Cholesterol is significantly redistributed in infected cells with strong colocalization with viral replication (Fig. 2S). However, with ATV pretreatment, cholesterol is more dispersed. Given that there are no therapeutics or vaccines against ZIKV and that statins are inexpensive with few side effects, they may be a valuable therapeutic against ZIKV.
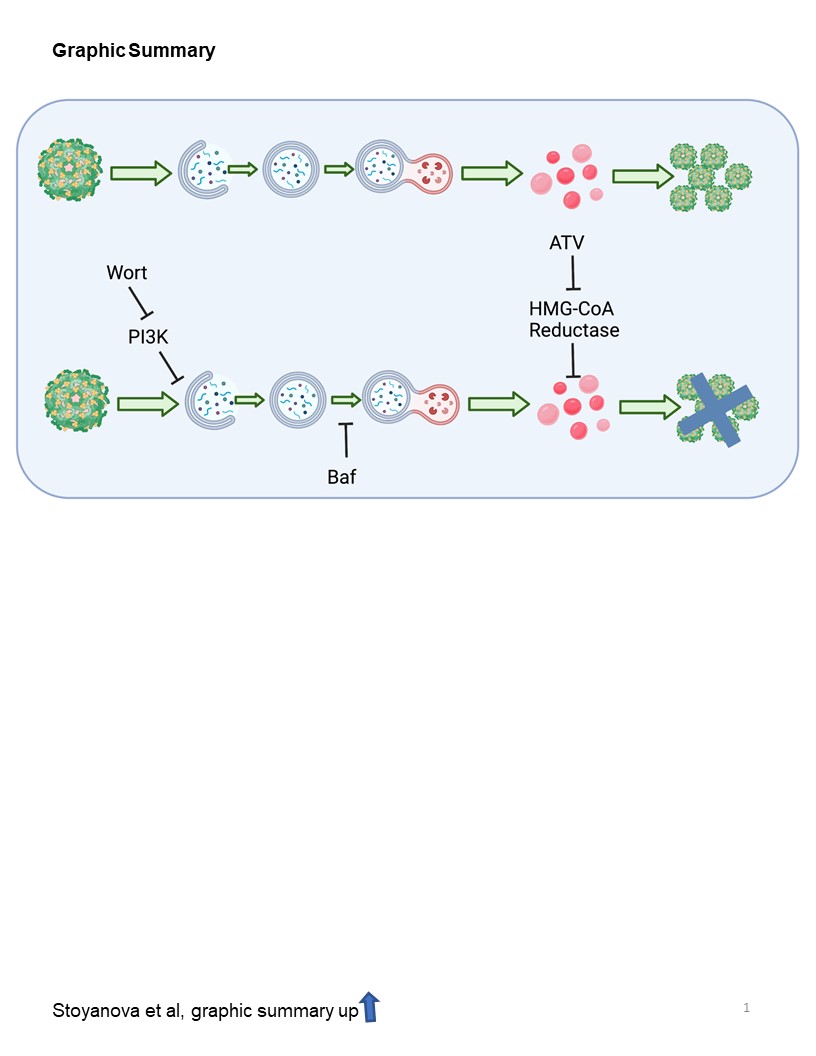
The interaction between ZIKV and the autophagic pathway is complex and may depend on cell type (19). In our model, autophagy positively contributes to LD accumulation and ZIKV infection as its inhibition reduces ZIKV E protein expression and synthesis of NS1 RNA (Fig. 3C-E). Previous studies have shown that ZIKV and other flaviviruses increase lipid droplet formation prior to the induction of autophagy (34, 35). Here we show that ZIKV increases autophagy as early as 6 hpi, prior to lipid droplet depletion, which according to our data occurs around 48 hpi (Fig. 3A). Wortmannin pretreatment inhibits early stages of autophagy, and like ATV, reduces LD availability in the cell and overall expression of the virus. Bafilomycin A1 treatment effectively abolished expression of ZIKV E protein (Fig. 4A, lower panel) while significantly decreasing LD (Fig. 4B) and redistributing host cholesterol (Fig. 2S). We also suggest that bafilomycin blocks cholesterol esterification (Fig. 3S) and this makes cholesterol unavailable for ZIKV-induced lipid increases, necessary to maintain viral replication. These results highlight the importance of the endosomal-lysosomal compartment for the manipulation of lipids required for the ZIKV lifecycle.
Flavivirus infection results in ER stress, which can promote autophagy and activate transcriptional changes related to the unfolded protein response (UPR). Induction of global ER stress in infected cells using tunicamycin led to almost twice as many lipids compared to Zika alone (Fig. 4S) However, Salubrinal, an inhibitor of eIF2α dephosphorylation and downregulator of the PERK branch of the UPR, did not change LD compared with ZIKV alone. Thus, global ER stress but not specifically the PERK pathway induces LD in infected cells. Future research should clarify the role of IRE1 and ATF6 pathways to LD in the context of ZIKV infection.
Taken together, there is a dynamic interplay between ZIKV and host lipids throughout the viral life cycle with autophagy contributing to Zika-induced LD accumulation as summarized in Fig. 5. Inhibition of either ER stress or autophagy alone suppresses, but does not eliminate, the amount of LD. Thus, it can be concluded that the availability of LD depends on several pathways. The induction of LD is a necessary component for replication of the virus, as inhibiting ZIKV-induced increase in lipids with ATV reduces replication (Fig. 6).
{kind=link}