Materials
Betaine was obtained from Sigma-Aldrich (St. Louis, MO, USA). Western blotting detection reagents were obtained from Amersham (Chalfont St. Giles, UK). Antibodies against β-actin, p-Akt, Akt, PPARγ, and ACC were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against FoxO6 and p-FoxO6 (Ser184) were obtained from Dr. H. Henry Dong (University of Pittsburgh, Pittsburgh, PA, USA). Anti-rabbit IgG-horseradish peroxidase-conjugated antibody and anti-mouse IgG-horseradish peroxidase-conjugated antibody were obtained from Amersham Pharmacia Biotech. Horseradish peroxidase-conjugated donkey anti-sheep/goat IgG was purchased from Serotec (Oxford, UK). Polyvinylidene difluoride (PVDF) membranes were obtained from the Millipore Corporation (Bedford, MA, USA). Dokdo-MARK™ protein size marker was obtained from ElpisBiotech (Daejeon, Korea).
Mouse Models
C57BLKS/J-lean and C57BLKS/J-db/db mice (8 weeks old, male) were purchased from Japan SLC (Hamamatsu, Japan). The mice were maintained under a 12 h light/dark cycle at 23 ± 1°C and 50 ± 5% relative humidity under specific pathogen-free conditions. Betaine was injected to db/db mice by oral gavage (50 mg/kg/day) for 3 weeks. Body weight and food intake were measured every other day. The mice were sacrificed after 3 weeks. All experiments were approved and performed in accordance with the regulations of the Chosun Univeristy Care and Used Committee (IACUC). All mice were housed at the Chosun University Specific pathogen free (SPF) Animal Care Unit.
Cell Culture System
HepG2 (human hepatocellular carcinoma) cells were obtained from the ATCC (American Type Culture Collection, Rockville, MD, USA). HepG2 cells were cultured in Dulbecco’s Modified Eagle Media (DMEM) (Nissui Co., Tokyo, Japan) supplemented with 10% heat-inactivated (56°C for 30 min) fetal bovine serum (Gibco, Grand Island, NY, USA), 233.6 mg/mL glutamine, 72 µg/mL penicillin streptomycin, and 0.25 µg/mL amphotericin B, and adjusted to pH 7.4–7.6 with NaHCO3 in a 5% CO2 atmosphere. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Western Blotting
Homogenized samples were boiled for 5 min in gel-loading buffer (125 mM Tris-Cl, 4% sodium dodecyl sulfate (SDS), 10% 2-mercaptoethanol, pH 6.8, 0.2% bromophenol blue) at a volume ratio of 1:1. Total protein-equivalents for each sample were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to PVDF membranes at 15 V for 1 h using a semi-dry transfer system. Membranes were immediately placed into a blocking buffer (1% non-fat milk) in 10 mM Tris, pH 7.5, 100 mM NaCl, and 0.1% Tween-20. Blots were allowed to block at room temperature for 1 h. Membranes were then incubated with specific primary antibodies at 4ºC overnight, followed by incubation with horseradish peroxidase-conjugated secondary antibodies at 25ºC for 1 h. Antibody labeling was detected by enhanced chemiluminescence according to the manufacturer's instructions. Pre-stained protein markers were used for measuring molecular weight.
Immunoprecipitation (Ip) Assay
Nuclear extracts were immunoprecipitated in a buffer containing 40 mM Tris-HCl (pH 7.6), 120 mM NaCl, 20 mM β-glycerophosphate, 20 mM NaF, 2 mM sodium orthovanadate, 5 mM EDTA, 1 mM PMSF, 0.1% NP40, leupeptin (2 µg/ml), aprotinin (1 µg/ml), and pepstatin A (1 µg/ml). Aliquots of cell extracts were centrifuged at 12,000 g at 4ºC for 15 min, incubated overnight at 4ºC with the correspondent antibody, and then incubated overnight at 4ºC with 50% protein A-agarose slurry. After washing the immunoprecipitates three times with immunoprecipitation buffer, the immunoprecipitated proteins were analyzed by SDS-PAGE and western blotting, as described above.
Serum Biochemical Analyses
Blood samples were collected from animals in each group after sacrifice. Serum samples were prepared by centrifugation (4℃, 2000 ⅹ g for 15min). ALT and AST levels were analyzed by using kits from Asan pharm (Asan, South Korea).
Rna Isolation And Real Time Rt-pcr
RNA isolation from liver (20 mg) or HepG2 cells (~ 2x106 cells) was performed using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Real-time polymerase chain reaction (RT-PCR) was performed to measure relative mRNA concentrations using the Roche LightCycler-RNA amplification kit (Roche Diagnostics, Indianapolis, IN, USA). All primers were obtained commercially from Integrated DNA Technologies (Coralville, IA, USA). The primer sequences are listed in Supplementary Table 1.
Luciferase Assays
PPARγ activities were estimated using an PPARγ-luciferase vector. Transfection was carried out using Lipofectamine 2000 (Invitrogen). Briefly, 1 × 104 cells per well were seeded in 48-well plates. When the cultured cells reached about 40% confluence, they were treated with DNA/Lipofectamine 2000 complexes (1 µg/µl) in 500 µl normal media (with 10% serum) for 24 h, then treated with the vector containing Akt (100 MOI), or FoxO6-siRNA (100 MOI) at 24 h after transfection. Subsequently, glucose (30 mM) was added for 8 h. Then, the cells were washed with PBS, and subjected to the Steady-Glo Luciferase Assay System (Promega, Madison, WI, USA). Luciferase activity was measured by a luminometer (GENious, TECAN, Salzburg, Austria).
Immunofluorescence
HepG2 cells were seeded at 1 × 103 cells per well in a 6-well plate, incubated for 24 h, fixed in 4% paraformaldehyde solution (15 min at room temperature), washed with PBS buffer, blocked with 3% normal goat serum (Gibco, Grand Island, USA), and immunostained using rabbit anti-PPARγ antibody (1:1000 dilution, Santa Cruz, CA) at 4°C overnight. Cells were then washed with TBS and incubated for 3 h in the presence of anti-rabbit IgG labeled with Alexa Fluor 488 (1:200; Invitrogen, CA, USA). Cell nuclei were visualized by immunostaining with Hoechst 33342 (1:1000; Invitrogen), and PPARγ was determined by confocal laser scanning microscopy (TCS SP2, Leica, Wetzler, Germany).
Lipid Content
Liver tissue and cell samples were homogenized in 400 µL of HPLC-grade acetone. After an overnight incubation with agitation at room temperature, 50 µL aliquots of acetone-extracted lipid suspensions were used to determine triglyceride concentrations via the Infinity triglyceride reagent (Thermo Electron). Liver lipid content was defined as mg of triglyceride per gram of total liver tissue or cellular proteins, as described earlier.
Histological Analysis
Oil Red O staining is used to identify cellular neutral lipid droplet accumulation. The cells were stained by the Oil Red O method. After treatments, the cells were washed three times with ice-cold PBS and fixed with 4% formalin for 30 min, and then dipped in 60% isopropanol for 5 min. After fixing, the cells were washed and stained with Oil Red O solution for 60 min at room temperature. After staining, the tissue or cells were washed three times with PBS to remove the unbound stain.
Livers were fixed in 10% neutral formalin and paraffin-embedded sections were stained with hematoxylin and eosin (H&E staining). The Oil Red O (Sigma, O1391) staining was done by described previously methods with optimal cutting temperature of frozen samples and consequent staining.
Cytotoxicity Assays
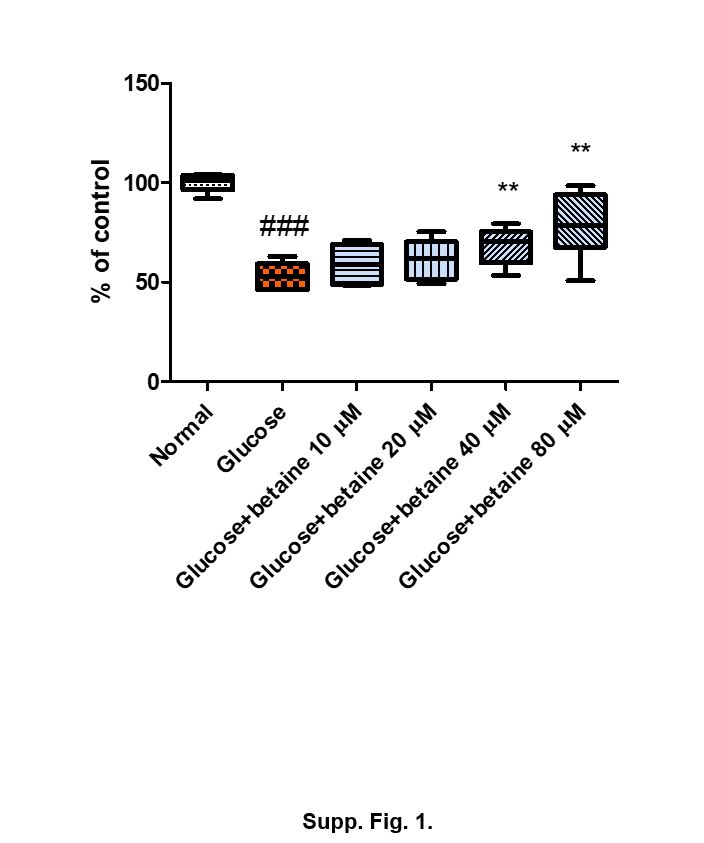
Cytotoxicity was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) from Aldrich Chemical Co. (Madison, WI). Cells were seeded into 96-well plates, incubated overnight for adherence, and subsequently exposed to 30 mM glucose with betaine for various periods of conc. At the end of the treatment, MTT reagent dissolved in PBS was added to the medium (final concentration of 0.5 mg/ml) and the plates were incubated in the dark for 1 h. After incubation, the supernatant was removed and formazan crystals were dissolved in 100 µl of dimethyl sulfoxide (DMSO) with gentle agitation. The absorbance in each well was measured at 450 nm by using a spectrophotometer.
Statistical Analysis
Data are expressed as the mean ± standard error means (SEM). GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA) was used for one-way ANOVA followed by Bonferroni post-tests, where p < 0.05 was considered statistically significant.
{kind=link}