2.1 Baseline characteristics of the study cohort
The demographic and clinical characteristics of patients are shown in Table 1. Patients with LAA and CE strokes showed a higher inflammatory state than the patients in the control group. hsCRP (LAA stroke vs. CE stroke vs. control: 4.57 ± 0.7 mg/L vs. 3.14 ± 0.92 mg/mL vs. 2.20 ± 0.42 mg/L) and leukocyte count (LAA vs. CE vs. control: 7.00 ± 0.26 × 109/L vs. 6.31 ± 0.32 × 109/L vs. 5.63 ± 0.23 × 109/L) significantly increased. In addition, D-dimer (LAA vs. CE vs. control: 0.99 ± 0.14 mg/L vs. 1.24 ± 0.25 mg/L vs. 0.47 ± 0.05 mg/L) showed a significant increase in stroke patients compared with asymptomatic controls (Fig. 2A).
2.2 Increased plasma TMAO and serum zonulin in patients with stroke
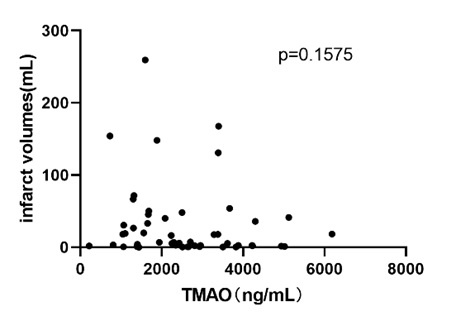
A total of 126 plasma samples (including stroke and control groups that met the inclusion criteria but did not provide fecal samples) were analyzed to study the plasma TMAO level difference between stroke patients and asymptomatic control. The results showed that patients with LAA and CE had significantly increased TMAO compared with asymptomatic controls (Fig. 2B) (TMAO: LAA stroke, 2931 ± 456.4 ng/mL vs. CE stroke, 4220 ± 577.6 ng/mL vs. control, 1663 ± 117.8 ng/mL). An additional 36 non-LAA and CE (stenosis < 50%) acute cerebral ischemia blood samples (Fig. 1) were included to study the difference between TMAO and the degree of vascular stenosis. The TMAO difference between patients with stroke with mild stenosis (stenosis < 50%) and moderate–severe stenosis (LAA, stenosis > 50%) was not significant. The TMAO of the mild stenosis (stenosis < 50%) acute cerebral ischemia group and the control group had no significant difference (Fig. 2C) (TMAO: LAA, 2292 ± 481.1 ng/mL vs. mild stenosis, 1892 ± 214.3 ng/mL vs. control, 1663 ± 117.8 ng/mL). Furthermore, the relationship between plasma TMAO and infarct volume or carotid plaque area in the stroke patients was studied. Although TMAO and infarct volume were not directly correlated (Supplementary Fig. S1), the plasma TMAO and carotid plaque area among patients with LAA (stenosis > 50%) were positively correlated (Fig. 2E; Spearman r = 0.333, P = 0.0093). However, in patients with acute ischemic stroke (< 50%), this correlation disappeared (Fig. 2F, P = 0.7252). The difference in serum zonulin levels between patients with stroke and the asymptomatic controls was analyzed using 161 serum samples (including stroke group or control group that met the inclusion criteria but did not have stool samples). As expected, patients with LAA and CE stroke showed significantly increased zonulin in serum compared with asymptomatic controls (Fig. 2D) (zonulin: LAA, 2.71 ± 1.17 ng/mL vs. CE, 2.17 ± 1.00 ng/mL vs. control, 0.43 ± 0.07 ng/mL).
Table 1
Demographic and clinical characteristics of the study participants
| | LAA | CE | Controls | P value |
| Blood samples Fecal samples Demographic | 85 (61 + 24*) 61 | 23 (20 + 3*) 20 | 59 (51 + 8*) 51 | |
| Age, years (mean) | 66 | 72 | 58 | < 0.05 |
| Gender (male/female) | 40/21 | 11/9 | 19/32 | < 0.05 |
| Habit | | | | |
| Drinking (yes/no) | 19/42 | 4/16 | 7/44 | |
| Smoking (yes/no) | 22/39 | 4/16 | 4/47 | |
| Hypertension (yes/no) | 42/19 | 16/4 | 15/36 | |
| T2DM (yes/no) | 24/37 | 3/17 | 4/47 | |
| Laboratory values | | | | |
| Neutrophilic granulocyte, 109/L | 4.80 ± 0.22 | 4.48 ± 0.34 | 3.41 ± 0.20 | < 0.05ab |
| ApoA I, g/L | 1.06 ± 0.27 | 1.16 ± 0.54 | 1.21 ± 0.04 | < 0.05a |
| ApoB, g/L | 0.97 ± 0.03 | 0.72 ± 0.053 | 0.87 ± 0.37 | < 0.05a |
| ApoAI/ApoB | 1.17 ± 0.06 | 1.66 ± 0.15 | 1.51 ± 0.09 | < 0.05a |
| HDL-C, mmol/L | 1.05 ± 0.032 | 1.12 ± 0.06 | 1.19 ± 0.04 | < 0.05a |
| LDL-C, mmol/L | 2.89 ± 0.12 | 2.21 ± 0.238 | 2.67 ± 0.78 | < 0.05b |
| Folic acid | 13.8 ± 1.27 | 16.83 ± 3.01 | 20.4 ± 2.2 | < 0.05a |
| GPDA, U/L | 83.18 ± 2.33 | 83.8 ± 3.33 | 98.6 ± 3.11 | < 0.05ab |
| α-HBDH, U/L | 155.68 ± 6.87 | 175.4 ± 16.73 | 140.24 ± 3.7 | < 0.05ab |
| Total cholesterol, mmol/L | 4.64 ± 0.13 | 3.84 ± 0.23 | 4.52 ± 0.12 | > 0.05 |
| Total bilirubin,µmol/L | 11.45(7.55) | 15.9(9.025) | 10.45(5.75) | < 0.05b |
| Direct bilirubin, µmol/L | 4(2.33) | 5.85(2.93) | 3.15(1.7) | < 0.05ab |
| γ-GT, U/L | 25(23.5) | 31(33.25) | 19.5(14.5) | < 0.05a |
| α-L-fucosidase, U/L | 19(4) | 21.5(10.25) | 20(6) | < 0.05b |
| β-HB, mmol/L | 0.08(0.19) | 0.07(0.07) | 0.05(0.03) | < 0.05a |
* Indicates blood specimens, including specimens that meet the inclusion criteria but do not have the fecal specimens needed for TMAO and zonulin analyses.
The tested indices with a normal distribution indicates mean ± SEM via one-way ANOVA. The indicator of disobedience is expressed as the median (interquartile range) and analyzed through a Kruskal–Wallis test. LAA indicates large artery atherosclerosis, CE denotes cardioembolism, HDL-C is high-density lipoprotein cholesterol, and LDL-C is low-density lipoprotein cholesterol. P > 0.05 means no significant statistical difference among the three groups. aP suggests a significant statistical difference between LAA and controls. bP implies a significant statistical difference between CE and controls. abP indicates a significant statistical difference among LAA, CE, and controls.
2.3 Alterations of gut microbiome composition in patients with stroke
2.3.1 Increased α Diversity In Laa And Ce
A total of 132 fecal samples from patients with LAA and CE and asymptomatic controls were analyzed. The species accumulation curve was used to determine whether the detected sample size was sufficient. The curve was smooth, and the sampling was sufficient; thus, data analysis can be performed (Fig. 3A). Rarefaction Curves and Chao indices were examined to characterize species richness. The results showed that the curves were nearly saturated in each group (Fig. 3B), and there was an increased trend in LAA compared with asymptomatic controls (Fig. 3C, P = 0.06318 for Chao richness). Species diversity and phylogeny were compared with other parameters, including Shannon and Simpson indices and phylogenetic diversity (PD) whole tree. The three ecological parameters showed that gut microbial diversity in the LAA group was much higher than that in the asymptomatic controls (Figs. 3D–3F). The species diversity between CE and the control increased according to the result of the Shannon indices and PD whole tree (Figs. 3G and 3H). This trend was also observed in the Simpson index (Fig. 3I, P = 0.063) but without statistical significance.
2.3.2 Increased β Diversity In Laa And Ce
The overall diversity in microbial composition was assessed via principal co-ordinates analysis (PCoA) and non-metric dimensional scaling (NMDS) analysis. Figures 4A–4B show that patients in the LAA group were more dispersed and had higher heterogeneity than those in the control group. On the basis of weighted UniFrac distance, significant differences were observed via PCoA and NMDS analysis between LAA and the control (Figs. 4A–4B, qualitative analysis by multi-response permutation procedure (MRPP), P = 0.002). The microbial communities between LAA and the control were significantly different for the unweighted UniFrac distance (MRPP, P = 0.01), Bray–Curtis distance, and Jaccard distance at the OUT level (Table 2). Although this trend was also observed when CE was compared with the control based on weighted UniFrac distance (Table 2, MRPP, P = 0.077), only unweighted UniFrac distance reached statistical significance (Figs. 4C–4D). Thus, LAA and CE may harbor a distinct gut microbial community composition compared with the control.
| Group | | A | Observed Delta | Expected Delta | P |
| LAA–control | Weighted_unifrac | 0.03535 | 0.25339 | 0.26268 | 0.002 |
| LAA–control | unWeighted_unifrac | 0.0078 | 0.50602 | 0.51 | 0.01 |
| LAA–control | Bray_curtis_OTU | 0.01071 | 0.71603 | 0.72378 | 0.001 |
| LAA–control | Jaccard_OTU | 0.00263 | 0.62787 | 0.62952 | 0.037 |
| CE–control | Weighted_unifrac | 0.01009 | 0.24088 | 0.24334 | 0.077 |
| CE–control | unWeighted_unifrac | 0.00847 | 0.51435 | 0.51875 | 0.026 |
Table 2: β diversity among LAA, CE, and control groups. A value > 0 indicates that the difference between groups is greater than the difference within the group. A value < 0 indicates the opposite. The smaller the value of ObserveDelta, the smaller the difference within the group; the smaller the ExpectDelta value, the smaller the difference between groups.
2.3.3 Altered Community Types In Laa And Ce
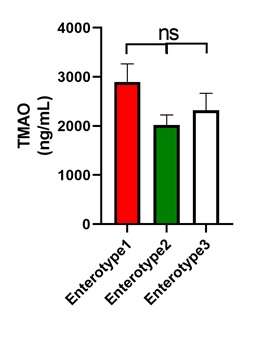
Microbial enterotype features were studied to investigate the shift in the gut microbiota community structure in the stroke patients and asymptomatic controls. Microbial enterotype features were calculated using the Jensen–Shannon distance of genera abundance and clustered samples with partitioning around medoids. A total of 132 samples were divided into three clusters (Fig. 5A). Enterotypes 1, 2, and 3 were dominated by Bacteroides, Prevotella, and Ruminococcus, respectively (Figs. 5B, P = 2e-16, P = 6.1e-16, and P = 0.0041, respectively; Kruskal–Wallis test). The association between enterotype distribution and stroke status was determined, and the results showed that patients with LAA were underrepresented in enterotype 2 (Prevotella) but overrepresented in enterotype 3 (Ruminococcus, Fig. 5C, Fisher’s exact test, P = 0.001999). CE stroke patients and the asymptomatic control exhibited a similar enterotype distribution (Figs. 5D–5F, P = 0.7536). Enterotype was investigated to determine the relationship between TMAO and gut microbiota. Previous research has revealed that the TMAO-producer phenotype should be identified [19], so we attempted to identify it based on enterotype stratification. The results showed that no significant difference existed among the three enterotypes (Supplementary Fig. 2). We failed to distinguish the TMAO producer phenotype via enterotype stratification.
2.3.4 Changed taxonomic composition profile in patients with LAA and CE
First, the taxonomic profile of gut microbiota in LAA was compared with that of the asymptomatic control individuals from the taxonomic composition plots. At the phylum level, Bacteroidetes, Firmicutes, Proteobacteria, Fusobacteria, and Actinobacteria represented more than 90% of the total bacterial community in the gut (Fig. 6A). Bacteroidetes and Firmicutes were the most dominant phyla in LAA and asymptomatic controls, and the changes observed between the two groups were significant at the phylum level. Bacteroidetes were significantly decreased in LAA, whereas Firmicutes, Proteobacteria, and Actinobacteria were significantly increased in LAA (Fig. 6A). When CE was compared with the asymptomatic control, significant changes were observed only in Bacteroidetes and Fusobacteria (Fig. 6B). A subsequent analysis of the microbiota relative abundances at other taxonomic levels (class, order, family, and genera) was performed. The 21 genera levels were significantly different between CE and the control. Among the genera, the relative abundances of Fusobacterium, Streptococcus, Dorea, Enterococcus, and Desulfovibrio were significantly increased in CE (top 20, Fig. 6C). Four genera, including Streptococcus, were significantly different at various taxonomic levels (Figs. 7A–7B; class, order, family, and genera). Twenty-seven genera were significantly different between LAA and the control (top 20, Fig. 6D). Parabacteroides, Klebsiella, and Lactobacillus relative abundances were significantly increased in LAA, and seven genera had obvious differences (Fig. 8; class, order, family, and genera).
2.3.5 Different Biomarkers Of Laa And Ce
Linear discriminant analysis effect size (LDA effect size) was performed on the fecal microbiota to identify the biomarkers of LAA, CE, and control. The differences in the taxa at different levels with a logarithmic LDA score > 2.0 and a P-value < 0.05 were considered. At the phylum level, Bacteroidetes was significantly enriched in the asymptomatic control, whereas Firmicutes and Proteobacteria were enriched in LAA. Significant differences were also observed at the genera level. The following eight bacterial taxa showed distinct relative abundances between the two groups in fecal microbiota: Collinsella, Parabacteroides, Alloprevotella, Lactobacillus, Anaerotruncus, Fastidiosipila, Mogibacterium, and Klebsiella (Figs. 9A–9B). Consistent with the results of the taxonomic composition profile (Fig. 6D), all of them were significant increased in LAA. A general linear model (GLM) [20] was designed to control the possible influences of age, gender, drinking, smoking, hypertension, and diabetes and further validate the results. The abundances of Parabacteroides and Fastidiosipila differed between the two groups (P = 1.33E-06 and P = 8.42E-06, Table 3).
Several species between CE and the control exhibited significant differences in abundance. At the phylum level, Bacteroidetes was the most enriched in the asymptomatic control, but Fusobacteria was significantly increased in CE. The following bacterial genera showed a distinct increase in relative abundances between CE and the control: Lactobacillus, Streptococcus, Peptostreptococcus, Fastidiosipila, Mogibacterium, and Fusobacterium (Figs. 10A–10B). The taxonomic composition profile produced the same result (Fig. 6C). Notably, after the possible confounding factors were adjusted (age, gender, drinking, smoking, hypertension, and diabetes), the Streptococcus abundance in the two groups was still statistically significant (P = 1.0286e-06, Table 3).
Table 3
Biomarkers of LAA, CE, and asymptomatic control after adjusting for possible confounding factors. Results of the GLMs for significant genera based on the group factors (LAA–asymptomatic controls or CE–asymptomatic controls) and possible confounding factors (age, gender, drinking and smoking habits, hypertension, and diabetes). Negative B values indicate that the taxa are associated with asymptomatic control, and positive numbers indicate that the taxa are associated with the LAA or CE. g means genera.
| Group | Names | B value | P value |
| LAA_Control | g__Parabacteroides | 18.32 | 1.33E-06 |
| LAA_Control | g__Fastidiosipila | 7.42 | 8.42E-06 |
| CE_Control | g__Streptococcus | 2.25 | 1.0286e-06 |
2.3.5.1 Gut microbiome-based signature-discriminated stroke and control
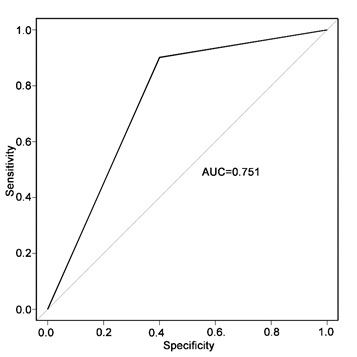
The potential of using gut microbiota as biomarkers for discriminating LAA and CE from control was assessed. First, Fastidiosipila and Parabacteroides were used separately to generate area under receiving operating characteristics (AUC) curves of 0.54 and 0.62, respectively (Fig. 11A). Second, multivariable stepwise logistic regression analysis [21] was applied to the list of LAA-associated genera identified by LEFSe to generate AUC. We found that the combination of eight genera, namely, Collinsella, Parabacteroides, Alloprevotella, Lactobacillus, Anaerotruncus, Fastidiosipila, Mogibacterium, and Klebsiella, could discriminate LAA from non-stroke controls effectively with an AUC of 0.843 (Fig. 11B). All diversity levels of microbiome identified by LEFSe (Fig. 9B) could significantly improve predictive performance (Fig. 11C; AUC, 0.931). The potential value between CE and the control was assessed, and Streptococcus generated an AUC of 0.7 (Fig. 11D). The combination of six bacterial taxa at the genus level could also effectively discriminate CE from non-stroke controls with an AUC of 0.762 (Fig. 11E). However, using all microbiomes identified by LEFSe (Fig. 10B) did not improve predictive performance (Supplementary Fig. 3; AUC, 0.751).
2.4 Role of baseline characteristic factors in the change in intestinal microbes
Studies have reported that microbiota composition changes with aging [22], and the present study is limited by the non-balanced age distribution of the included patients and controls (P > 0.05, Table 1). Therefore, whether age factors account for the main factors in the alterations of intestinal microbes in patients with stroke was explored. A subgroup analysis was performed on 75 samples with a balanced age distribution for further analysis (Supplementary Table 1). Consistent with the aforementioned analysis results, a significant difference was observed between the two groups (group 1: stroke and group 2: control) in the α and β diversity analyses (Figs. 12A–12D). The gut microbiota of the control group can be separated from that of the patient group based on the weighted UniFrac matrix (P = 0.001, Figs. 12A–12B).
Additionally, although HTN and T2DM are widely known to be connected with gut microbiome dysfunction, we did not exclude stroke patients or control with comorbidities (HTN and T2DM) to reflect the real signature of clinical practice. Nineteen non-diabetic, hypertensive stroke patients (including LAA and CE) and 32 controls without diabetes and hypertension were screened to evaluate the disrupted patterns of gut microbiome resulting mainly from stroke (Supplementary Table 2; BS, Stroke; Con, control). The age distributions were well balanced between the two groups (P = 0.257, independent t-test). The α and β diversity indices showed significant differences between BS and Con (P = 0.018, Figs. 13A–13F). The gut microbiota of patients (non-diabetic, HTN, and control) could be separated from that of others (non-diabetic, HTN, and stroke group) via PCoA and NMDS (Figs. 13A–13B). The subgroup analyses replicated the previous results obtained after adjusting for possible confounding factors (Table 3). This finding indicates that Parabacteroides can still serve as a marker of stroke (Table 4). Consequently, the difference in gut microbiome between the stroke and control groups was mainly due to stroke.
Existing literature shows that age has a considerable influence on TMAO. The plasma TMAO between the two groups was analyzed after age equalization. The TMAO levels increased significantly in patients with stroke (Fig. 14; LAA, 2763 ± 408.7 ng/mL vs. control, 1642 ± 144.1 ng/mL). The relationship between intestinal microbes and stroke was also analyzed. Fecal samples of patients with stroke were analyzed according to the location of vascular stenosis (internal carotid and vertebrobasilar arteries), and the location of cerebral infarction (brain stem, cerebellar, and other parts of infarction). The difference between α and β diversity indices was not significant (results are not shown), indicating that the changes in intestinal microbes are not associated with the location of the infarct nor the location of vascular stenosis.
| Biomarker | LDA | P |
| p__Bacteroidetes.c__Bacteroidia.o__Bacteroidales.f__Porphyromonadaceae | 3.1 | 0.009 |
| p__Bacteroidetes.c__Bacteroidia.o__Bacteroidales.f__Porphyromonadaceae.g__Parabacteroides | 2.85 | 0.024 |
Table 4. Biomarker between stroke patients and the control after adjusting for confounding factors HTN and T2DM. P, phylum; c, class; o, order; f, family; g, genera; and s, species.
2.5 Relationship Between Bacteria And Laboratory Or Clinical Indices
The relationship between bacteria in the genera detected in all samples and clinical indices was explored. Several genera played catalytic roles in the pathogenesis of stroke. We found that certain microorganisms had high correlation with multiple clinical indicators (Fig. 15A). Parabacteroides, a discriminative bacteria detected by LEFSe, could be regarded as a biomarker in LAA. Parabacteroides was positively correlated with infarct volume (measured using DWI) and carotid artery plaque area (measured using carotid duplex ultrasound), negatively correlated with ADL, and positively correlated with serum neutrophilic granulocyte, creatine kinase, and total cholesterol. It was also positively correlated with zonulin, which exhibited a significant increase in patients with LAA and CE. Further exploration revealed that the association of Parabacteroides can be attributed to P. distasonis (Fig. 15B) and Parabacteroides_sp._HGS0025 (Fig. 15C). The abundance of Alistipes also showed a positive correlation with infract volumes, carotid artery plaque area, and creatine kinase and a negative correlation with ADL. The abundance of Alistipes and other genera (Anaerotruncus, Odoribacter, and Ruminococcus) exhibited a significant correlation with TMAO. Streptococcus, as a biomarker in CE, showed a positive correlation with fasting blood glucose but a negative correlation with total cholesterol. Notably, several genera levels may have played an opposite role in the course of stroke. Haemophilus was negatively correlated with infract volume and positively correlated with the ADL score. Haemophilus was significantly elevated in the control group compared with the LAA group (Fig. 15D), indicating that these genera may play roles in preventing stroke.
2.6 Alterations in gut microbiome function in patients with stroke
The functional and metabolic changes in the microbial communities in LAA, CE, and the controls were studied by inferring the metagenomes from the 16sRNA data and predicting the potential function of the gut microbiota using PICRUSt. The predicted functional categories were based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologue (KO). In the level 2 KEGG pathway, amino acids, glycan, secondary metabolites, terpenoids, polyketides, cofactors, and vitamins were significantly low in the LAA microbiome. Membrane transport, signal transduction, and xenobiotic biodegradation and metabolism were high in LAA (Fig. 16B). In the level 3 KEGG pathway (Fig. 16A), unsaturated fatty acids, ketone body biosynthesis, fatty acid, and glycerolipid metabolism were significantly increased in the LAA microbiome. Excessive accumulation of extracellular glutamate, a principal excitatory neurotransmitter, is a major factor that contributes to the death of ischemic penumbra [23]. Several genes associated with D-glutamine and D-glutamate metabolism had low representation in the microbiome of LAA compared with the control. In addition, the functional modules related to bacterial invasion of epithelial cells were significantly increased in the LAA microbiome, and combined with the elevated serum zonulin, these may indicate an increased intestinal permeability in patients with LAA. Several genes related to antigen processing and presentation and several immunology pathways on innate immunity, such as the peroxisome proliferator-activated and nucleotide-binding oligomerization domain (NOD)-like receptor signaling pathways, were significantly decreased in the LAA microbiome (Fig. 16A). Furthermore, secondary metabolites associated with antibiotics, such as biosynthesis of vancomycin group antibiotics and streptomycin, were significantly decreased in the LAA microbiome. Such metabolites may promote the growth of pathogenic microorganisms and change the gut microbiome composition in patients with stroke.
The functional difference between CE and the control was compared. In the level 2 KEGG pathway (Fig. 17A), the microbial gene functions related to membrane transport were significantly higher in CE than in the control. Inversely, cofactors, vitamins, glycan biosynthesis, and metabolism were significantly decreased in CE. The top 20 differences in the level 3 KEGG pathway are shown in Fig. 17C. Consistent with the functional changes in LAA, glycerolipid metabolism was significantly high, and amino sugar, nucleotide sugar metabolism, and protein digestion and absorption were significantly low in CE. In the function of the LAA microbiome, the NOD-like receptor signaling pathway and several secondary metabolites associated with antibiotics, such as streptomycin and vancomycin, also decreased in CE (Fig. 17B).
The levels of circulating TMAO were significantly increased in the stroke patients. The relationship between the microbiome function and TMAO in the study cohort was studied. In the microbiota of LAA patients, the genes involved in the synthesis, transport, and metabolism of methylamine-containing source nutrients [24] were significantly increased (Table 5). Examples of the genes include lipopolysaccharide cholinephosphotransferase (K07271), choline (K00108, K02168), betaine (K00130, K03762, K05020), phosphatidylcholine (K01004), carnitine (K08279), L-carnitine (K05245), and TMA (K14084, K14083). Gut microbial enzyme complexes are involved in the formation of primary metabolite TMA. Carnitine can be converted by microbiota into TMA via carnitine TMA lyases, such as CntA/CntB. Choline can be converted into TMA via choline TMA lyases, such as CutC/CutD, and TMAO can be converted to TMA via TMAO reductase. From the results of the PICRUSt analysis, we could not detect the KOs linked to the first four enzymes, namely, K20038 for choline trimethylamine-lyase CutC, K20037 for choline trimethylamine-lyase activating enzyme CutD, K22443 for carnitine monooxygenase subunit CntA, and K22444 for carnitine monooxygenase subunit CntB. Several KOs could be defined to TMAO reductase (table.6). Among such KOs, K07811, K07821, and K03532 were significantly higher in the LAA microbiome compared with the control (Fig. 18A). Searching the NCBI nucleotide database by using the phrase “TMAO reductase” showed that Enterobacteriaceae (family), Helicobacter (genus), and many other human intestinal origin bacteria can reduce TMAO to TMA [25]. The results of the present work indicate that the relative abundance of Enterobacteriaceae and Helicobacter increased more significantly in the LAA microbiome than in the control (Fig. 18B). LEFSe analysis indicated that Enterobacteriaceae was significantly increased in patients with stroke and may be considered a biomarker (Table 7). Thus, the microorganisms in the LAA microbiome can convert more dietary sources of TMAO to TMA than those in the control. This phenomenon may partly contribute to the increased plasma TMAO levels in LAA. We noticed that such KOs could be projected onto the same metabolic pathway, that is, K000680 methane metabolism (Fig. 19, part of methanogenesis, TMAO as a substrate). Methane metabolism was significantly higher in LAA than in the control (Fig. 12B), but other information about the pathway that we could obtain from PICRUSt analysis was limited. Metagenomics sequencing technology was necessary for further research.
Table 5
KOs involved in the synthesis, transport, and metabolism of TMA-containing source nutrient (searched from KEGG, https://www.kegg.jp/) * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
| Entry | Name | Definition | P |
| K00108 | betA, CHDH | choline dehydrogenase | 0.003 |
| K00130 | betB, gbsA | betaine-aldehyde dehydrogenase | 0.014 |
| K01004 | pcs | phosphatidylcholine synthase | 0.037 |
| K02168 | betT, betS | choline/glycine/proline betaine transport protein | 0.041 |
| K03762 | proP | proline/betaine transporter | 0.024 |
| K05020 | opuD, betL | glycine betaine transporter | 0.008 |
| K05245 | caiT | L-carnitine/gamma-butyrobetaine antiporter | 0.034 |
| K07271 | licD | lipopolysaccharide cholinephosphotransferase [EC:2.7.8.-] | 1.27E-05 |
| K08279 | caiE | carnitine operon protein CaiE | 0.044 |
| K14083 | mttB | trimethylamine—corrinoid protein Co-methyltransferase [EC:2.1.1.250] | 9.83E-04 |
| K14084 | mttC | trimethylamine corrinoid protein | 0.043 |
Table 6
KOs involved in TMAO reductase (searched from KEGG, https://www.kegg.jp/).
| Entry | Name | Definition |
| K07811 | torA | trimethylamine-N-oxide reductase (cytochrome c) [EC:1.7.2.3] |
| K07821 | torY | trimethylamine-N-oxide reductase (cytochrome c), cytochrome c-type subunit TorY |
| K07812 | torZ | trimethylamine-N-oxide reductase (cytochrome c) [EC:1.7.2.3] |
| K03532 | torC | trimethylamine-N-oxide reductase (cytochrome c), cytochrome c-type subunit TorC |
| K03533 | torD | TorA specific chaperone |
| Biomarker | LDA | P |
| p__Proteobacteria | 3.23 | 0.00 |
| p__Proteobacteria.c__Gammaproteobacteria | 3.19 | 0.02 |
| p__Proteobacteria.c__Gammaproteobacteria.o__Enterobacteriales | 3.21 | 0.01 |
| p__Proteobacteria.c__Gammaproteobacteria.o__Enterobacteriales.f__Enterobacteriaceae | 3.21 | 0.01 |
| p__Proteobacteria.c__Gammaproteobacteria.o__Enterobacteriales.f__Enterobacteriaceae.g__Klebsiella | 2.95 | 0.04 |
| Table 7. Biomarkers in patients with stroke. P, phylum; c, class; o, order; f, family; g, genera. |
11413148: K18277(ttm); 1.5.8.2: K00317(dmd-tmd) ;1.5.8.1: K00317 (dmd-tmd)
17.2.3: K07811 (torA), K03532 (torC), K03533 (torD), K07812 (torZ), K07821 (torY)
MtbA: K14082(mtbA) MttB: K14083(mttB) MttC: K14084(mttC)
MtbA: K14082(mtbA) MtbB: K16178(mtbB) MtbC: K16179(mtbC)
MtbA: K14082(mtbA) MtmB: K16176(mtmB) MtmC: K16177(mtmC)
{kind=link}
{kind=link}
{kind=link}