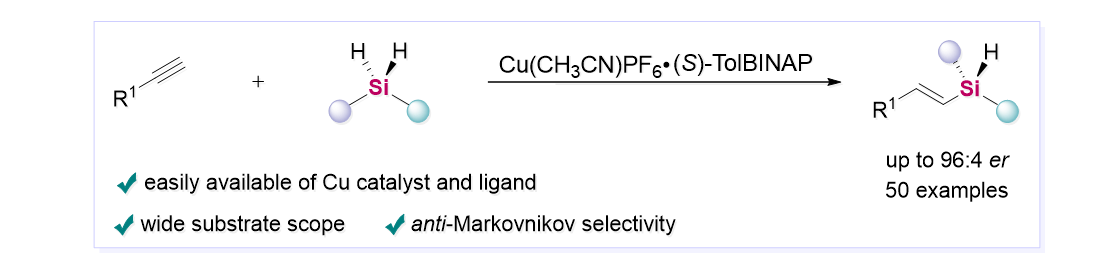
Herein we report an unprecedented copper-catalyzed asymmetric anti-Markovnikov hydrosilylation of alkynes with dihydrosilanes for the synthesis of silicon-stereogenic alkenyl silanes in good regio- and enantioselectivities. Demonstrated by a diverse collection of substrates, the process grants access to chiral alkenylhydrosilanes with a simple Cu/(S)-Tol-BINAP catalytic system. The origin of the enantiocontrol was investigated by density functional theory (DFT) calculations, which revealed a quite complicated scenario involving multiple pairs of diastereomeric transition structures accounting for the high enantiocontrol under the seemingly simple conditions.
Article
Enantioselective Construction of Si-Stereogenic Linear Alkenylhy-drosilanes via Copper-Catalyzed Anti-Markovnikov Hydrosilyla-tion of Alkynes
https://doi.org/10.21203/rs.3.rs-2324096/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
In comparison with the synthesis of C-stereogenic compounds, so far only limited strategies have been developed for the synthesis of chiral organosilicon compounds featuring Si-stereogenic centers. Due to the substantially longer C-Si bond counteracts the formation of compact transition states, the control of construction of chiral silicon-stereocenters becomes more challenging.1 Notably, organosilicon compounds are important in various areas such as bioactive molecules and organic materials.2-8 However, chiral organosilicon compounds do not exist in nature. Therefore, asymmetric catalytic synthesis of silicon-stereogenic silanes is highly desirable in organic chemistry. Traditional methods for the synthesis of Si-stereogenic silanes rely on use of stoichiometric amounts of chiral auxiliaries or resolution processes.9-16 During recent decades, impressively, transition metal-catalyzed desymmetrization of dihydrosilanes and tetraoganosilanes have provided an alternative access to construct the silicon-stereogenic center.17-21 In fact, the asymmetric catalytic transformation of dihydrosilanes has attracted considerable attention because the obtained chiral product retains a reactive Si-H bond may offer more possibilities for further functionalization.22-54 The popular methods of intermolecular desymmetrization of dihydrosilanes to generate silicon-stereogenic silanes include: 1) hydrosilylation of alkenes27-29, 1,3-dienes30, alkynes31-33 and ketones34-36; 2) carbenes insertion37-39; 3) arylation and dehydrogenative coupling reactions40-44; 4) alcoholysis45-54 etc (Scheme 1a).
Among them, the asymmetric intermolecular hydrosilylation of unsaturated C-C bonds has emerged as a powerful and atom-economic tool for the stereochemical control of chirality at silicon. In 2012, the Tomooka group reported the first example for catalytic enantioselective synthesis of alkenylhydrosilane based on Pt(0)-catalyzed hydrosilylation of internal alkynes with Taddol-derived phosphite ligand.31 Later, Huang and co-workers developed a CoCl2/PyBox complex catalyzed hydrosilylation to form the Si-stereogenic compounds with excellent enantiomeric excess.32 Recently, Xu and co-workers achieved a Pd-catalyzed hydrosilylation of ynones with binaphthyl-phosphoramidite ligand to construct chiral silanes with moderate regioselectivities and good enantioselectivities.33 The examples reported by Hou27, Xu28 and He29 groups for the synthesis of chiral silicon-stereogenic centers from alkenes and dihydrosilanes required chiral half-sandwich scandium catalyst and rhodium catalyst, respectively. Very recently, Meng and co-workers described a Co(acac)2/(S,S)-Chiraphos catalyzed asymmetric hydrosilylation of conjugated 1,3-dienes for simultaneous construction of carbon- and silicon-stereogenic centers.30 Despite great advances achieved in this area, there is room for the development of other earth abundant metals catalyzed general methods to expand the variety of products. It is worth noting that so far only two cases for anti-Markovnikov selective hydrosilylation of unsaturated C-C bonds were presented for preparing the linear alkyl substituted Si-stereogenic silanes with terminal olefins as substrates.27,29 On the basis of previous studies on copper-catalyzed asymmetric hydrosilylation of unsaturated C-C bonds,55-60 herein we reported a copper-catalyzed regio- and enantioselective hydrosilylation of terminal alkynes to synthesize the silicon-stereogenic linear alkenylhydrosilanes (Scheme 1b).
Table 1. Development of reaction conditions. a

a Conditions: 1a (0.2 mmol), 2a (0.3 mmol), copper catalyst (5 mol %) and ligand (5 mol %) in extra dry solvent (0.2 M) was stirred at 50 °C for 15 h under argon atmosphere. b Yield was determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard. c The er was determined by HPLC. d THF or 2-MeTHF/m-xylene [1:4 (v/v)] as solvent. e Cu(CH3CN)PF6·L2 complex as catalyst. f 3 mol % Cu(CH3CN)PF6·L2 complex. g Isolated yield.
Evaluation of reaction conditions. On the basis of our previously reported Cu-catalyzed anti-Markovnikov hydrosilylation of alkynes,61 the investigation was initiated with using 4-ethynyl-1,1'-biphenyl 1a and (2,6-dimethoxyphenyl)(methyl)silane 2a (Table 1). The reaction was conducted at 50 oC for 15 h in THF with Cu(CH3CN)4PF6 as catalyst and (S)-BINAP as ligand. The desired product 3aa was obtained in 56% yield with 92:8 er (entry 1). Inspired by this preliminary result, we then tested several Cu catalysts such as Cu(CH3CN)4BF4, Cu(CH3CN)4OTf, CuI and Cu(OAc)2 to promote this reaction. Unfortunately, the reaction catalyzed by other copper catalysts did not generate the desired product. Then, other commercially available bisphosphine ligands were screened (entries 6-12). The reaction proceeded smoothly with (S)-Tol-BINAP (L2) as ligand and afforded the desired product with 94:6 er. But a bulkier ligand (S)-Xyl-BINAP (L3) was examined and provided insufficient yield and enantioselectivity. A higher enantioselectivity (95:5 to 99:1 er) could be observed in the presence of (R)-DTBM-SEGPHOS (L5), (R)-tBu-MeOBIPHEP (L6) or (R,R)-Ph-BPE (L7) as ligand, but the bulky and electron-rich bisphosphine ligands cannot maintain high yields (4-24%) of 3aa. Furthermore, various solvents were investigated for this hydrosilylation reaction catalyzed by Cu(CH3CN)4PF6. Replacement of THF with 2-MeTHF could increase the yield (72%), whereas a slightly decreased enantiomeric ratio (91:9) was obtained (entry 13). When this reaction was conducted in PhOMe, toluene, or m-xylene, the desired product was obtained in low yield but with good er values (entries 14-16). Thus, the mixed solvents were tried, and the mixture of THF and m-xylene [1:4 (v/v)] furnished the product 3aa in 52% yield with 95:5 er (entry 17). When THF was replaced by 2-MeTHF as solvent in mixed system, the desired product was formed in 60% yield with 95:5 er (entry 18). Additionally, when a complex Cu(CH3CN)PF6·L2 as precatalyst instead of Cu(CH3CN)4PF6 and L2 was used to ensure efficient generation of the catalytic species and a precise ratio of ligand and copper, the reaction gave 3aa in 66% yield without loss of enantioselectivity. Moreover, decreasing the catalyst loading had no obvious change on the yield or enantioselectivity. The absolute configuration of (S)-3aa was determined by X-ray diffraction analysis (CCDC: 2183555).
Scope of the reaction. After establishing the optimized reaction conditions, we turned to evaluate the substrate scope and limitation of this Cu-catalyzed asymmetric hydrosilylation of alkyne (Scheme 2). A broad range of aryl substituted alkynes (1a-z, 1a′-j′) were applicable under the reaction condition with moderate to good yields and enantioselectivities. Simple phenylacetylene was converted to styrenylhydrosilane (3ba) in 66% yield with 94:6 er. Different alkyl substituents installed on phenyl ring did not affect the reaction efficiency and enantioselectivities, affording the chiral organosilane products in 56-77% yield with 93:7-95:5 er (3ca–3fa). Other electron-donating groups such as –OMe (1g), –SMe (1h), –OBn (1i) and –N(Ph)2 (1j) on phenyl ring were all well tolerated to provide the corresponding products in 54-79% yields with 93:7-94:6 er. In addition, the substrates with electron-withdrawing substituents were also investigated. It was found that the substrate with 4-fluoro, 2-chloro, 3-chloro or 4-bromo halide group at phenyl ring still worked well with dihydrosilane (2a) and furnished the desired products (3ka-3na). When the 4-CO2Me-substituted phenylacetylene was applied in this reaction, the product (3oa) was delivered in 50% yield with a slightly diminished enantioselectivity. Next, other aryl rings
such as thienyl, naphthyl, and 9H-fluorenyl substituted terminal alkynes were tested to react with dihydrosilane 2a, all took place smoothly to afford the corresponding products (3pa, 3qa, and 3ra) in good results, respectively. Interestingly, excellent chemoselectivity was observed when the internal alkynyl and terminal alkenyl group substituted phenylacetylenes were subjected to this transformation, the products 3sa and 3ta were formed with good enantioselectivities. Moreover, reaction of the conjugated enynes 1u and 1v with 2a under the optimized reaction conditions afforded the linear conjugated dienylsilane products (3ua, 3va) in 55% and 71% yield with 93:7 and 92:8 er, respectively. Other aliphatic groups substituted alkynes displaying various functional substituents, such as carbonyl (1w), ester (1x and 1y), chloride (1z), and ether (1a′, 1b′) were all well tolerated, providing the desired products (3wa-3b′a) with good to excellent yields and enantioselectivities. It was noted that pure aliphatic chain substituted alkynes (1c′ and 1d′) were also suitable substrates and converted to the desired products in 68% and 65% yields with the same 92:8 er.
To demonstrate the synthetic potential and diversity of this methodology, we applied the reaction to late-stage functionalization of natural products, bioactive molecules, pharmaceutical and material building blocks, such as β-estradiol (3e′a), (-)-borneol (3f′a), geraniol (3g′a), estrone (3h′a), D-menthol (3i′a), and cholestanol (3j′a). An array of chiral Si-stereogenic alkenylhydrosilanes were obtained in moderate to good yields with excellent enantioselectivities, regardless of existing diverse functional groups or complex molecular structures.
Furthermore, to investigate the possible roles of the 2,6-dimethoxy functional group in substrates 2 in controlling the enantioselectivity and/or yield, a series of dihydrosilanes 2b–2o were synthesized and subjected to this reaction. Simple methylphenylsilane (2b) and n-pentylphenylsilane (2c) generally led to products with eroded enantioselectivities but maintained moderate yields. More bulky substituents on aryl ring of the silane could slightly improve the enantioselectivities (2d–2j). Notably, mesityl(methyl)silane (2h) led to an increased enantioselectivity of product 3ah (91:9 er), albeit with a decreased yield. A series of silanes with electron-donating (2k–2n) and electron-withdrawing (2o) groups were explored to examine the electronic effect. The results revealed that dimethoxy functional group in dihydrosilane 2a not only has steric effect to control the enantioselectivities and yields, but also has an electron-donating effect favor this transformation.
Synthetic transformations of compound 3a′a. Finally, the reaction was performed on a gram scale, which gave alkenylhydrosilane 3a′a in 73% yield with 93:7 er. To further demonstrated the utility of Si-stereogenic vinylhydrosilanes, several transformations of 3a′a were performed. Oxidation of the Si-stereogenic monohydrosilane 3a′a with m-CPBA provided the corresponding chiral silanol 4 in 75% yield with a decreased enantioselectivity.62 Then, hydrosilylation of 3a′a could smoothly convert it to 5 in 68% yield without loss of enantiopurity in the presence of [Rh(cod)Cl]2.39 In addition, hydrogenation of 5 successfully delivered silane 6 in a good yield with 93:7 er via a hydroboration/protodeboronation strategy.63
Computational studies of enantioselectivity controlled. To elucidate the origin of the enantiocontrol, we resorted to DFT calculation of the only step involving generation of the stereocenter, i.e., the metathesis between the incipient styrenyl Cu(I) and the silane. We systematically sampled the conformational space by including all permutations of the major flexible elements in the interaction of alkenyl Cu(I) with silane, namely, (i) the relative position of silane and styrenyl moiety around the tetrahedral Cu(I) center (denoted as 1 or 2 respectively), (ii) the s-cis or s-trans orientation of the styrenyl moiety relative to the C‒P bond projecting toward the silane (denoted as a or b respectively), (iii) orientation of the silane phenyl opposite to or toward the styrenyl Cu(I) (denoted as c or d respectively), and (iv) engagement of the pro-(R) or pro-(S) silane hydride (denoted as S or R respectively) (Figure S1). Such consideration leads to altogether 16 transition structures whose geometries were located and their Gibbs free energies determined using the double hybrid functional PWPB95,64 with Grimme’s DFT-D3(BJ) empirical dispersion65 and the SMD implicit solvation model66 (Figure 1a). The calculations suggest that the conformers having the styrenyl moiety at one side (Set 1) generally favor the observed (S)-configuration, while the structures in the other set (Set 2) all favor the minor (R)-antipode. Importantly, the former set of conformers includes several members that are appreciably lower in energy and are thus dominating in population. Quantitatively, an 85:15 er is theoretically predicted (Table S5), agreeing reasonably well with the experimental observation. For a comparison, calculations with three typical hybrid functionals M0667, PBE068 and ω-B97XD69 with DFT-D3 empirical dispersion all agree with the five low-lying conformers within 2 kcal/mol range accountable for the enantiocontrol, while they differ in the order of the relative energy of a couple of them (Figure S3). According to the PWPB95-D3(BJ) functional, 1ad_S (62%), 1ac_S (17%) and 1bd_R (7%) are the most dominant. Inspection of the geometries of the five lowest-lying transition structures (Figure 1B) shows no obvious correlation between the key bond length parameters with the relative stability of the structures, so distortion interaction analyses70 were performed to probe the origin of the energy difference (Figure 1B). First, comparison of the most dominant TS, 1ad_S and its “epimer” 1ad_R suggest that the major factor contributing to the energy difference is the appreciably larger distortion energy of the silane fragment in the latter, likely rising from shorter C‒Si bond (2.19Å vs 2.27Å) and slightly longer Si‒H bond (1.61Å vs 1.60Å). Interaction region indicator (IRI) analysis71 (processed by Multiwfn 3.8 dev.72) revealed the appreciable differences in non-covalent interaction regions: the former features a larger and stronger van der Waals (vdW) attraction region between the Si‒Me moiety and one of the P-Ar of the ligand as compared to the latter (Scheme 1C, red circled part in i vs ii), which likely helps increase the interaction energy, while reduce the strain of the fragments. Besides this dominant pair, 1ac_S, having the phenyl moiety of the silane being stacked with the P-Ar of the other side of the styrenyl moiety, also makes non-negligible contribution (17%) to the formation of the major enantiomer. In contrast, its “epimer”, 1ac_R, is 4.33 kcal/mol higher in energy and makes almost no contribution, due also to the much larger distortion energy of the silane moiety (Si-H bond length: 1.79 Å vs 1.73 Å in 1ac_S) being not sufficiently counterbalanced by the interaction energy. The opposite is observed for the least-contributing pair (1bd_S vs 1bd_R). Here 1bd_R is slightly lower in energy (0.26 kcal/mol) despite of its larger distortion energies from both fragments, as this effect is offset by the larger interaction energy, resulting in a later yet lower-energy transition state. For the latter two pairs, differences in the interaction region diagrams are non-obvious (Figure S4).
In conclusion, we have developed a copper-catalyzed asymmetric anti-Markovnikov hydrosilylation of alkynes to construct Si-stereogenic alkenylhydrosilanes using a simple catalytic system, employing commercially available (S)-Tol-BINAP as ligand. This efficient method enables a scalable and robust hydrosilylation of various alkynes in good to high yields and enantioselectivities under mild conditions. The products can be further elaborated to Si-chiral silanol and silanes. DFT computational studies elucidated the origin of the enantiocontrol, suggesting that more than one pairs of diastereomeric transition structures are responsible for the observed high enantioncontrol despite of the deceptively simple reaction conditions. Distortion interaction analyses and interaction region indicator (IRI) analyses revealed the key differences in the key transition structures.
Data availability
Source data are provided with this paper. The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author on request.
Acknowledgment
We gratefully acknowledge research support of this work by the funding of the National Natural Science Foundation of China (21871240), the State Key Laboratory of Elemento-organic Chemistry Nankai University (202001), and the Fundamental Research Funds for the Central Universities (WK2060000017), the Strategic Priority Research Program of the CAS (XDPB14).
Author contributions
Y.-H. Xu directed the project and composed the manuscript with revisions provided by the other authors. J.-L. Xu and Z.-L Wang performed the experiments. J.-B. Zhao performed the DFT calculations. All the authors were involved the analysis of results and discussions of the project.
Competing interests
The authors declare no competing interests.
- Weickgenannt, A. & Oestreich, M. The Renaissance of Silicon Stereogenic Silanes: a Personal Account. Asymmetric Synth. II, 35 – 42 (2013).
- Tacke, R. & Linoh, H. Bioorganosilicon Chemistry. Organic Silicon Compounds; J. Wiley and Sons: New York, 1989.
- Franz, A. K. & Wilson, S. O. Organosilicon Molecules with Medicinal Applications. J. Med. Chem. 56, 388–405 (2013).
- Fujii, S. & Hashimoto, Y. Progress in the Medicinal Chemistry of Silicon: C/Si Exchange and Beyond. Future Med. Chem. 9, 485–505 (2017).
- Ramesh, R. & Reddy, D. S. Quest for Novel Chemical Entities through Incorporation of Silicon in Drug Scaffolds. J. Med. Chem. 61, 3779–3798 (2018).
- Remond, E., Martin, C., Martinez, J. & Cavelier, F. Silicon-Containing Amino Acids: Synthetic Aspects, Conformational Studies, and Applications to Bioactive Peptides. Chem. Rev. 116, 11654–11684 (2016).
- Magnus, P. Silicon in Organic, Organometallic, and Polymer Chemistry; J. Wiley and Sons: New York, 2000.
- Kawakami, Y., Kakihana, Y., Ooi, O., Oishi, M., Suzuki, K., Shinke, S. & Uenishi, K. Control of Stereochemical Structures of Silicon-Containing Polymeric System. Polym. Int. 58, 279–284 (2009).
- Sommer, L. H. Stereochemistry, Mechanism and Silicon; an Introduction to the Dynamic Stereochemistry and Reaction Mechanisms of Silicon Centers; McGraw-Hill: New York, 1965.
- Strohmann, C., H€ornig, J. & Auer, D. Synthesis of a Highly Enantiomerically Enriched Silyllithium Compound. Chem. Commun. 766 – 767 (2002).
- Trzoss, M., Shao, J. & Bienz, S. Preparation of a ‘Si-centered’ Chiral Auxiliary by Resolution. Tetrahedron: Asymmetry 15, 1501–1505 (2004).
- Rendler, S., Auer, G. & Oestreich, M. Kinetic Resolution of Chiral Secondary Alcohols by Dehydrogenative Coupling with Recyclable Silicon-Stereogenic Silanes. Angew. Chem. Int. Ed. 44, 7620–7624 (2005).
- Rendler, S., Auer, G., Keller, M. & Oestreich, M. Preparation of a Privileged Silicon-Stereogenic Silane: Classical versus Kinetic Resolution. Adv. Synth. Catal. 348, 1171–1182 (2006).
- Rendler, S., Oestreich, M., Butts, C. P. & Lloyd-Jones, G. C. Intermolecular Chirality Transfer from Silicon to Carbon: Interrogation of the Two-Silicon Cycle for Pd-Catalyzed Hydrosilylation by Stereoisotopochemical Crossover. J. Am. Chem. Soc. 129, 502–503 (2007).
- Igawa, K., Takada, J., Shimono, T. & Tomooka, K. Enantioselective Synthesis of Silanol. J. Am. Chem. Soc. 130, 16132–16133 (2008).
- Igawa, K., Kokan, N. & Tomooka, K. Symmetric Synthesis of Chiral Silacarboxylic Acids and Their Ester Derivatives. Angew. Chem. Int. Ed. 49, 728–731 (2010).
- Xu, L-W., Li, L., Lai, G.-Q. & Jiang, J.-X. The Recent Synthesis and Application of Silicon-Stereogenic Silanes: A Renewed and Significant Challenge in Asymmetric Synthesis. Chem. Soc. Rev. 40, 1777–1790 (2011).
- Shintani, R. Recent Advances in the Transition-Metal-Catalyzed Enantioselective Synthesis of Silicon-Stereogenic Organosilanes. Asian J. Org. Chem. 4, 510–514 (2015).
- Wu, Y. & Wang. P. Silicon-Stereogenic Monohydrosilane: Synthesis and Applications. Angew. Chem. Int. Ed. e202205382 (2022).
- Yuan, W. & He, C. Enantioselective C–H Functionalization toward Silicon-Stereogenic Silanes. Synthesis 54, 1939–1950 (2022).
- Shintani, R. Recent Progress in Catalytic Enantioselective Desymmetrization of Prochiral Organosilanes for the Synthesis of Silicon-Stereogenic Compounds. Synlett 29, 388–396 (2018).
- Huang, Y.-H., Wu, Y., Zhu, Z., Zheng, S., Ye, Z., Peng, Q. & Wang, P. Enantioselective Synthesis of Silicon-Stereogenic Monohydrosilanes by Rhodium-Catalyzed Intramolecular Hydrosilylation. Angew. Chem. Int. Ed. 134, e202113052 (2022).
- Guo, Y., Liu, M.-M., Zhu, X., Zhu, L. & He, C. Catalytic Asymmetric Synthesis of Silicon-Stereogenic Dihydrodibenzosilines: Silicon Central-to-Axial Chirality Relay. Angew. Chem. Int. Ed. 133, 14006–14010 (2021).
- Ma, W., Liu, L. C., An, K., He, T. & He, W. (2021). Rhodium-Catalyzed Synthesis of Chiral Monohydrosilanes by Intramolecular C – H Functionalization of Dihydrosilanes. Angew. Chem. Int. Ed. 60, 4245–4251.
- Chen, S., Mu, D., Mai, P.-L., Ke, J., Li, Y. & He, C. Enantioselective Construction of Six- and Seven-Membered Triorgano-Substituted Silicon-Stereogenic Heterocycles. Nat. Commun. 12, 1249 (2021).
- Yuan, W., You, L., Lin, W., Ke, J., Li, Y. & He, C. Asymmetric Synthesis of Silicon-Stereogenic Monohydrosilanes by Dehydrogenative C–H Silylation. Org. Lett. 23, 1367–1372 (2021).
- Zhan, G., Teng, H. L., Luo, Y., Lou, S. J., Nishiura, M. & Hou, Z. Enantioselective Construction of Silicon-Stereogenic Silanes by Scandium-Catalyzed Intermolecular Alkene Hydrosilylation. Angew. Chem. Int. Ed. 57, 12342–12346 (2018).
- Zhao, Z.-Y., Nie, Y.-X., Tang, R.-H., Yin, G.-W., Cao, J., Xu, Z., Cui, Y.-M., Zheng, Z.-J. & Xu, L.-W. Enantioselective Rhodium-Catalyzed Desymmetric Hydrosilylation of Cyclopropenes. ACS Catal. 9, 9110–9116 (2019).
- He, T., Liu, L.-C., Ma, W.-P., Li, B., Zhang, Q.-W. & He, W. Enantioselective Construction of Si-Stereogenic Center via Rhodium-Catalyzed Intermolecular Hydrosilylation of Alkene. Chem. Eur. J. 26, 17011–17015 (2020).
- Wang, L., Lu, W., Zhang, J., Chong, Q. & Meng, F. Cobalt-Catalyzed Regio-, Diastereo- and Enantioselective Intermolecular Hydrosilylation of 1,3-Dienes with Prochiral Silanes. Angew. Chem. Int. Ed. e202205624 (2022).
- Igawa, K., Yoshihiro, D., Ichikawa, N., Kokan, N. & Tomooka, K. Catalytic Enantioselective Synthesis of Alkenylhydrosilanes. Angew. Chem. Int. Ed. 51, 12745–12748 (2012).
- Wen, H., Wan, X. & Huang, Z. Asymmetric Synthesis of Silicon-Stereogenic Vinylhydrosilanes by Cobalt-Catalyzed Regio- and Enantioselective Alkyne Hydrosilylation with Dihydrosilanes. Angew. Chem. Int. Ed. 57, 6319–6323 (2018).
- Xie, J.-L., Xu, Z., Zhou, H.-Q., Nie, Y.-X., Cao, J., Yin, G.-W., Bouillon, J.-P. & Xu, L.-W. Palladium-Catalyzed Hydrosilylation of Ynones to Access Silicon-Stereogenic Silylenones by Stereospecific Sromatic Interaction-Assisted Si-H Activation. Sci. China. Chem. 64, 761–769 (2021).
- Corriu, R. J. P. & Moreau, J. J. E. Asymmetric Hydrosilylation of Ketones Catalysed by a Chiral Rhodium Complex. J. Organomet. Chem. 64, C51–C54 (1974).
- Hayashi, T., Yamamoto, K. & Kumada, M. Asymmetric Synthesis of Bifunctional Organosilicon Compounds via Hydrosilylation. Tetrahedron Lett. 15, 331–554 (1974).
- Ohta, T., Ito, M., Tsuneto, A. & Takaya, H. Asymmetric Synthesis of Silanes with a Stereogenic Centre at Silicon via Hydrosilylation of Symmetric Ketones with Prochiral Diaryl Silanes Catalysed by Binap-RhI Complexes. J. Chem. Soc., Chem. Commun. 2525–2526 (1994).
- Yasutomi, Y., Suematsu, H. & Katsuki, T. Iridium(III)-Catalyzed Enantioselective Si-H Bond Insertion and Formation of an Enantioenriched Silicon Center. J. Am. Chem. Soc. 132, 4510–4511 (2010).
- Nakagawa, Y., Chanthamath, S., Fujisawa, I., Shibatomi, K. & Iwasa, S. Ru(II)-Pheox-Catalyzed Si–H Insertion Reaction: Construction of Enantioenriched Carbon and Silicon Centers. Chem. Commun. 53, 3753–3756 (2017).
- Jagannathan, J. R., Fettinger, J. C., Shaw, J. T. & Franz, A. K. Enantioselective Si – H Insertion Reactions of Diarylcarbenes for the Synthesis of Silicon-Stereogenic Silanes. J. Am. Chem. Soc. 142, 11674–11679 (2020).
- Kurihara, Y., Nishikawa, M., Yamanoi, Y. & Nishihara, H. Synthesis of Optically Active Tertiary Silanes via Pd-Catalyzed Enantioselective Arylation of Secondary Silanes. Chem. Commun. 48, 11564–11566 (2012).
- Chen, L., Huang, J.-B., Xu, Z., Zhang, Z.-J., Yang, K.-F., Cui, Y.-M., Cao, J. & Xu, L.-W. Palladium-Catalyzed Si–C Bond-Forming Silylation of Aryl Iodides with Hydrosilanes: An Enhanced Enantioselective Synthesis of Silicon-Stereogenic Silanes by Desymmetrization. RSC Adv. 6, 67113–67117 (2016).
- Koga, S., Ueki, S., Shimada, M., Ishii, R., Kurihara, Y., Yamanoi, Y., Yuasa, J., Kawai, T., Uchida, T., Iwamura, M., Nozaki, K. & Nishihara, H. Access to Chiral Silicon Centers for Application to Circularly Polarized Luminescence Materials. J. Org. Chem. 82, 6108–6117 (2017).
- Chen, S., Zhu, J., Ke, J., Li, Y. & He, C. Enantioselective Intermolecular C – H Silylation of Heteroarenes for the Synthesis of Acyclic Si-Stereogenic Silanes. Angew. Chem. Int. Ed. e202117820 (2022).
- Mu, D., Pan, S., Wang, X., Liao, X., Huang, Y. & Chen, J. Enantioselective Synthesis of Acyclic Monohydrosilanes by Steric Hindrance Assisted C–H Silylation. Chem. Commun. 58, 7388–7391 (2022).
- Corriu, R. J. P. & Moreau, J. J. E. Synthese Asymetrique D'alcoxysilanes Catalysee par des Complexes du Rhodium. Tetrahedron Lett. 45, 4469–4472 (1973).
- Corriu, R. J. P. & Moreau, J. J. E. Asymmetric Synthesis at Silicon: II. Alcoholysis of Prochiral Organosilicon Compounds Catalysed by Rhodium Complexes. J. Organomet. Chem. 120, 337–346 (1976).
- Schmidt, D. R., OMalley, S. J. & Leighton, J. L. Catalytic Asymmetric Silane Alcoholysis: Practical Access to Chiral Silanes. J. Am. Chem. Soc. 125, 1190–1991 (2003).
- Xu, J.-X., Chen, M.-Y., Zheng, Z.-J., Cao, J., Xu, Z., Cui, Y.-M. & Xu, L.-W. Platinum-Catalyzed Multicomponent Alcoholysis/Hydrosilylation and Bis-hydrosilylation of Alkynes with Dihydrosilanes. ChemCatChem 9, 3111 – 3116 (2017).
- Long, P.-W., Bai, X.-F., Ye, F., Li, L., Xu, Z., Yang, K.-F., Cui, Y.-M., Zheng, Z.-J. & Xu, L.-W. Construction of Six-Membered Silacyclic Skeletons via Platinum-Catalyzed Tandem Hydrosilylation/Cyclization with Dihydrosilanes. Adv. Synth. Catal. 360, 2825–2830 (2018).
- Zhu, J., Chen, S. & He, C. Catalytic Enantioselective Dehydrogenative Si – O Coupling to Access Chiroptical Silicon-Stereogenic Siloxanes and Alkoxysilanes. J. Am. Chem. Soc. 143, 5301–5307 (2021).
- Zhu, J. & He, C. Catalytic Enantioselective Synthesis of Silicon-Stereogenic Alkoxysilanes and Siloxanes. Synlett 32, 1575–1580 (2021).
- Yang, W., Liu, L., Guo, J., Wang, S.-G., Zhang, J.-Y., Fan, L.-W., Tian, Y., Wang, L.-L., Luan, C., Li, Z.-L., He, C., Wang, X., Gu, Q.-S. & Liu, X.-Y. Enantioselective Hydroxylation of Dihydrosilanes to Si-Chiral Silanols Catalyzed by In Situ Generated Copper(II) Species. Angew. Chem. Int. Ed. e202205743 (2022).
- Yuan, W., Zhu, X., Xu, Y. & He, C. Synthesis of Si-Stereogenic Silanols by Catalytic Asymmetric Hydrolytic Oxidation. Angew. Chem. Int. Ed. e202204912 (2022).
- Gao, J., Mai, P.-L., Ge, Y., Yuan, W., Li, Y. & He, C. Copper-Catalyzed Desymmetrization of Prochiral Silanediols to Silicon-Stereogenic Silanols. ACS Catal. 12, 8476–8483 (2022).
- Gribble Jr, M. W., Pirnot, M. T., Bandar, J. S., Liu, R. Y. & Buchwald, S. L. Asymmetric Copper Hydride-Catalyzed Markovnikov Hydrosilylation of Vinylarenes and Vinyl heterocycles. J. Am. Chem. Soc. 139, 2192–2195 (2017).
- Nishino, S., Hirano, K. & Miura, M. Cu-Catalyzed Reductive gem-Difunctionalization of Terminal Alkynes via Hydrosilylation/Hydroamination Cascade: Concise Synthesis of α-Aminosilanes. Chem. Eur. J. 26, 8725–8728 (2020).
- Xu-Xu, Q.-F., Yang, P., Zhang, X. & You, S.-L. Enantioselective Synthesis of 4-Silyl-1,2,3,4-tetrahydroquinolines via Copper(I) Hydride Catalyzed Asymmetric Hydrosilylation of 1,2-Dihydroquinolines. Synlett 32, 505–510 (2021).
- Xu, J.-L., Xu, Z.-Y., Wang, Z.-L., Ma, W.-W., Sun, X.-Y., Fu, Y. & Xu, Y.-H. Copper-Catalyzed Regiodivergent and Enantioselective Hydrosilylation of Allenes. J. Am. Chem. Soc. 144, 5535–5542 (2022).
- Li, S., Xu, J.-L. & Xu, Y.-H. Copper-Catalyzed Enantioselective Hydrosilylation of Allenes to Access Axially Chiral (Cyclohexylidene)ethyl Silanes. Org. Lett. 24, 6054–6059 (2022).
- Zhang, M., Ji, Y., Zhang, Z. & Zhang, C. Copper-Catalyzed Highly Selective Hydrosilylation of Silyl or Boryl Alkene: A Method for Preparing Chiral Geminated Disilyl and Borylsilyl Reagents. Org. Lett. 24, 2756–2761 (2022).
- Wang, Z.-L., Zhang, F.-L., Xu, J.-L., Shan, C.-C., Zhao, M. & Xu, Y.-H. Copper-Catalyzed Anti-Markovnikov Hydrosilylation of Terminal Alkynes. Org. Lett. 22, 7735–7742 (2020).
- Hillenbrand, J., Leutzsch, M., Yiannakas, E., Gordon, C. P., Wille, C., Nöthling, N., Copéret, C. & Fürstner, A. “Canopy Catalysts” for Alkyne Metathesis: Molybdenum Alkylidyne Complexes with a Tripodal Ligand Framework. J. Am. Chem. Soc. 142, 11279–11294 (2020).
- Ding, W. & Song, Q. Chemoselective Catalytic Reduction of Conjugated α,β-Unsaturated Ketones to Saturated Ketones via a Hydroboration/Protodeboronation Strategy. Org. Chem. Front. 3, 14–18 (2016).
- Goerigk, L. & Grimme, S. Efficient and Accurate Double-Hybrid Density Functionals-Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics and Noncovalent Interactions. J. Chem. Theo. Compt. 7, 291–309 (2011).
- Grimme, S., Ehrilich, S. & Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Compt. Chem. 32, 1456–1465 (2011).
- Marenich, A. V., Cramer, C. J. & Truhlar. D. G. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
- Zhao, Y. & Truhlar, D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 120, 215–241 (2008).
- Adamo, C. & Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 110, 6158–6169 (1999).
- Chai, J.-D. & Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
- Bickelhaupt, F. M. & Houk, K. N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed. 56, 10070–10086 (2017).
- Lu, T. & Chen, Q. Interaction Region Indicator (IRI): A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. Chemistry-Methods 1, 231–239 (2021).
- Lu, T. & Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 33, 580–592 (2012).
Schemes 1-3 are available in the Supplementary Files section.
There is NO Competing Interest.
- xjlSI.pdf
- Scheme13.docx
Scheme 1. Approaches for the synthesis of silicon-stereogenic monohydrosilanes. Scheme 2. Exploration of substrate scope.a, b, c Scheme 3. Gram-scale synthesis and transformations of chiral silane products.
- GraphicalAbstract.png
{kind=link}