1. Experimental animals
6-8-week-old male APN-KO mice (weight 18-20 g) were obtained from professor Bian Yunfei, Second Hospital of Shanxi Medical University. 6-8-week-old male C57BL/6 mice (weight 18-20g) were obtained from Animal Center of Shanxi Medical University. The mice with free diet were placed in a suitable temperature and humidity environment throughout the experiment. All procedures related to animals in this study were approved by the Ethics Committee of Shanxi Medical University, and followed the People’ s Republic of China’ s Guidelines for the Care and Use of Laboratory Animals.
2. Agarose gel electrophoresis
The genotypes of APN-KO mice were identified by agarose gel electrophoresis. The mouse tail tissue about 5 × 10-3 m was put into the EP tube, then the tail lysate was added, and put into the 55 ℃ constant temperature water bath for the night. The DNA of mice tail tissue was extracted and amplified, and the primer sequences were as follows: P1: GGCTCTCTGGGAGAGGCGAG, P2: CCATCACGGCCTGGTGTGCC, P3: TTCGCCATTCAGGCTGCGCA. The samples were agarose gel electrophoresis, after electrophoresis, the glue was placed in the automatic exposure instrument.
3. Establishment of β1-AA actively immunized model
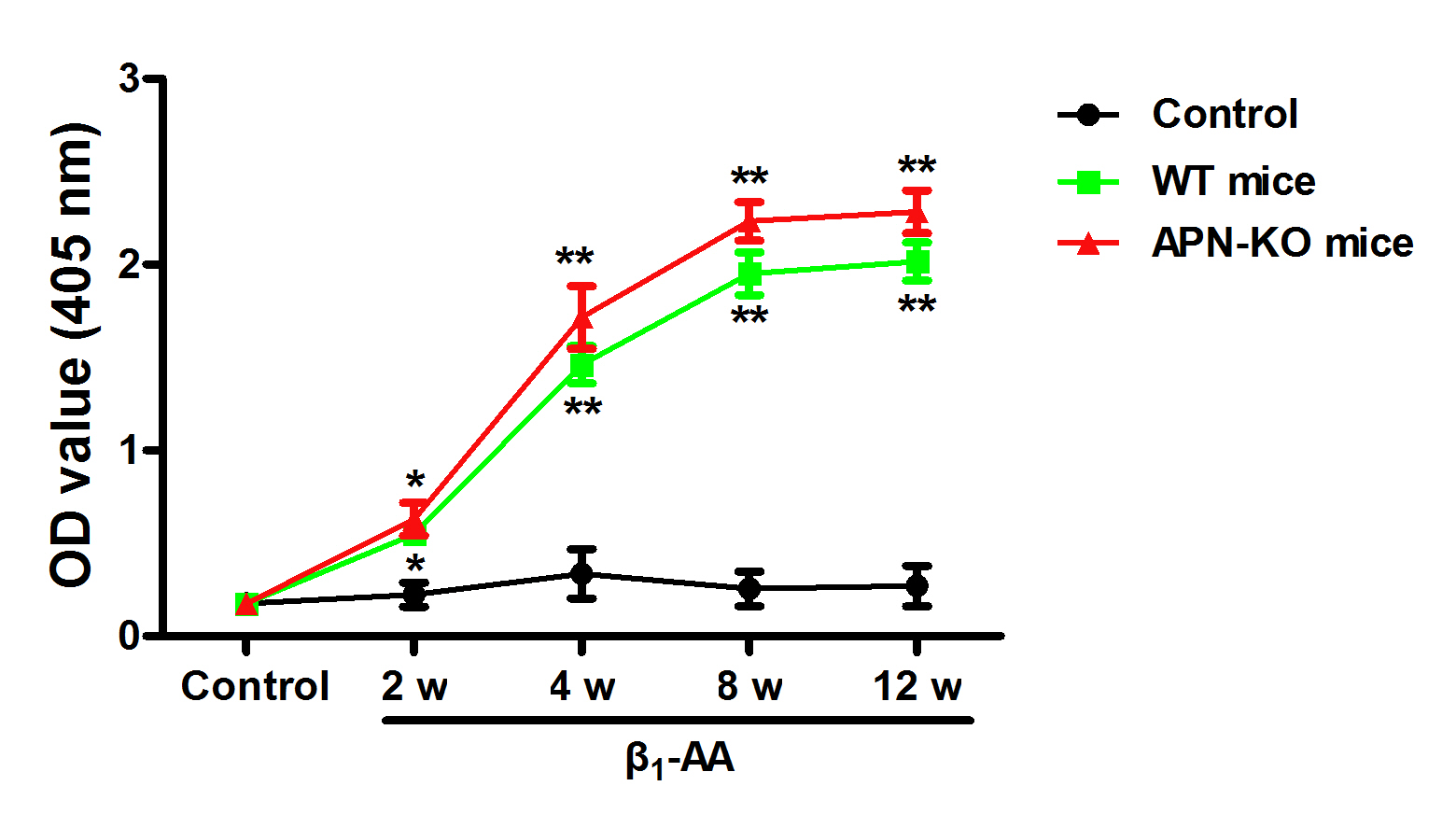
6-8-week-old male APN-KO mice and WT (C57BL/6) mice were randomly divided into actively immunized group and solvent control group, with 8 mice in each group. The peptide of β1-AR-ECII (GLS, Shanghai, Chinese) was dissolved and diluted with Na2CO3 solution (100 mM, pH 11.0), and then mixed with Freund’s complete adjuvan (Sigma-Aldrich, USA). After 1:1 emulsification and mixing, the mice were immunized with multiple injected subcutaneously into the back (0.4 μg/g) during the first immunization. Subsequently, diluted peptide of β1-AR-ECII emulsified with incomplete Freund's adjuvant (Sigma-Aldrich, USA), and single subcutaneous injection was used to strengthen immunization once every 2 weeks for 12 weeks. In the solvent control group, the same amount of Na2CO3 solution was used to replace the antigen solution (Supplementary Figure).
4. SA-ELISA
SA-ELISA was used to detect the level of β1-AA in the serum of actively immunized mice described previously[5]. The 96 well plates was used for antigen coating, and blank control group, solvent control group, positive control and actively immunized group were set up. β1-AR-ECII was dissolved in Na2CO3 (100 mM,PH=11.0) to prepare a solution with the final concentration of 10 μg/ml, and 50 μl was added into each well. Except for the blank control group, the other groups were coated with antigen at 4 °C overnight. The wells were saturated with PMT (1% (w/v) bovine serum albumin, 0.1% (v/v) Tween 20 in PBS, PH 7.4). The solvent control group, positive control, blank control and serum to be tested were diluted and added to 96 well plates. Then, the 96 well plates was added with biotinylated antibody (Zhongshan Golden Bridge Biotechnology, 1:2000) and horseradish peroxidase streptavidin (Zhongshan Golden Bridge Biotechnology, 1:2000) in 96 well plates. The above steps were respectively incubated at 37 °C for 1 h. The substrate (2.5 mM H2O2, 2 mM 2, 2'-azinodi (ethylbenzthiazoline) sulfuric salt (ABTS, Bio Basic Inc., AD0002, Markham, ON, Canada)) was added for 30 min. Finally, the 96 well plates was put into the microplate reader, and the OD value of each group was measured at 405 nm.
5. Affinity chromatography
First, a β1-AA-positive animal model was established by actively immunized rats with β1-AR-ECII, as described in the previous studies[16]. Then, animal serum from actively immunized rats and the control group were collected and extracted using MAbTrap Kit (GE Healthcare, Uppsala, Sweden) for affinity and purification of IgG.
6. Cell culture
H9c2 cells were purchased from Chinese Academy of Sciences Cell Bank (Shanghai, China). H9c2 cells were cultured in a complete medium containing 10% fetal bovine serum (Sijiqing, Shanghai, China) and 100 U/ml penicillin and 100 μg/ml streptomycin (Solarbio, P1400-100, Beijing, China), and were incubated at 37 °C in a humidified atmosphere of 5% CO2. The cells were subcultured every other day. Cell grouping and treatment: control group (treated with 1 μM negative IgG for 24 h), β1-AA group (treated with 1 μM β1-AA for 24 h), β1-AA + adiponectin group (450-27, pepro tech, USA) (pretreated with 10 μg/l adiponectin for 1 h and then treated with 1 μM β1-AA for 24 h), β1-AA + adiponectin + Compound C (s7840, Selleck, USA) (pretreated with 20 μM Compound C for 30 min, then 10 μg/l adiponectin was added for 1 h before adding 1 μM β1-AA for 24 h), β1-AA + Compound C group (pretreated with 20 μM Compound C for 30 min and then added 1 μM β1-AA for 24 h).
7. Western Blotting
Western blotting was used to detect the protein expression levels of LC3B (ab48394, Abcam, 1:1000), p62 (ab56416, Abcam, 1:1000), AMPK (#5831, CST, 1:1000), p-AMPK (#2535, CST, 1:1000) in each group. Samples of each group were collected and added with RIPA lysis buffer (p0013b, beyond biotechnology, China) for protein extraction. BCA Kit (23225, Thermo Scientific, Rockford, IL, USA) was used to determine the protein concentration, and then boiled to denaturate. The protein was analyzed by SDS-PAGE assay (the sample volume was 40 μg) and transfered to the PVDF membrane (IPVH00010, Millipore, Billerica, MA, USA). Then blocking for 2 h in 5% non-fat dry milk which was dissolved with 1 × Tris-buffered saline and incubated with the corresponding antibodies at 4 °C overnight. TBST was used to wash the membranes, then the membranes were incubated with the corresponding secondary antibodies(zb-2305, zb-2301, Zhongshan Golden Bridge biotechnology, 1:1000) at room temperature. After washing the membrane with TBST, the membranes were placed in the automatic exposure instrument. After adding Super ECL Plus (Applygen Technologies ), the blots were exposed and the gray value was analyzed by Image J. GAPDH was used as internal references to calculate the relative expression of different proteins.
8. Real-time PCR
We detected the mRNA levels of autophagy related genes Beclin1 and LC3B by Real-time PCR. Firstly, total RNA was extracted by Trizol method, then the total RNA was reverse transcribed to cDNA. We used SYBR Green Kit (Takara, Japan) to amplify. The specific sequence of primers were as follows: Beclin1 (Gen-Bank ID NM_001034117.1), upstream primer: 5′-GAAACTGGACACGAGCTTCAAGA-3′, downstream primer: 5′-ACCATCCTGGCGAGTTTCAATA-3′. LC3B (Gen-Bank ID NM_022867.2), upstream primer: 5′-AGCTCTGAAGGCAACAGCAACA-3′, downstream primer: 5′-GCTCCATGCAGGTAGCAGGAA -3′. GAPDH (Gen-Bank ID NM_017008.3), upstream primer: 5′-GGCACAGTCAAGGCTGAGAATG-3′, downstream primer: 5′-ATGGTGGTGAAGACGCCAGTA -3′. GAPDH was selected as the internal control. Data was quantified by the relative quantitative 2-ΔΔCt method.
9. CCK-8
Cell viability was measured with a cell counting kit-8(CCK-8). The H9c2 cells were seeded on 96 well plates (2 × 109/l). After H9c2 cells adhered to the wall, the H9c2 cells were pretreated with 5 μg/l, 10 μg/l and 30 μg/l adiponectin for 1 h before adding 1 μM β1-AA for 24 h. Then, 10 μl CCK-8 reagent (CK04, Dojindo Molecular Technologies, Kumamoto, Japan) was added into each well. When the color of the solution changes to brown yellow, the absorbance values of each group at 450 nm were detected by microplate reader. According to the absorbance value of each group, the cell survival rate of each experimental group compared with the control group was calculated. viability %= [(AS−AB)/(AC−AB)]×100%, where AS is the absorbance of the samples with β1-AA, AC is the absorbance of the DMEM media, and AB is the absorbance of the control.
10. Small animal ultrasound
The changes of cardiac function in mice were detected by small animal ultrasound. A small animal anesthesia machine was used for gaseous anesthesia, then put the mice into the anesthesia box and wait for the mice to faint. Fix the anesthetized mice on the mouse plate with adhesive tape, and maintain anesthesia by inhalation, remove the hair in the precordial area of the mice with depilatory creams, and gently wipe the residual hair with three distilled water to avoid causing artifacts. Fix the mice on the ultrasound platform, and smear the coupling agent on the precordial area of the mice. The left ventricular ejection fraction (EF%), Fractional shortening (FS%), LV internal diameter at end systole (LVID (s)) and LV internal diameter at end diastole (LVID (d)) were measured by M-mode ultrasound in the short axis section of the heart. After the detection, the precordial coupling agent was wiped off and put back into the cage.
11. Masson trichrome staining
Masson trichrome staining was used to observe the degree of myocardial fibrosis in mice. At the 12 week of immunization, mice were killed. The remaining blood in the heart was pumped out in PBS and fixed in 4% paraformaldehyde. The fixed tissue was washed with water and embedded in paraffin and sectioned (4 μm). After dewaxing, the tissue was stained with masson trichrome stain kit (Solarbio, Beijing, China) and sealed with neutral gum. All tissue slides were analyzed by optical microscopy (Olympus, BX45, Olympus Corporation, Tokyo, Japan) by the double-blind fashion.
12. Statistical analysis
Data are expressed as means ± SD. Statistical analysis was performed with SPSS software (version 16.0, SPSS Inc., Chicago, IL, USA). Two independent sample tests were used to compare the means of two independent samples and one-way ANOVA was applied after a Bonferroni post hoc test for more than two samples. P < 0.05 were considered statistically significant.
{kind=link}