Boutique shRNA screens identified known and novel regulators of breast cancer growth.
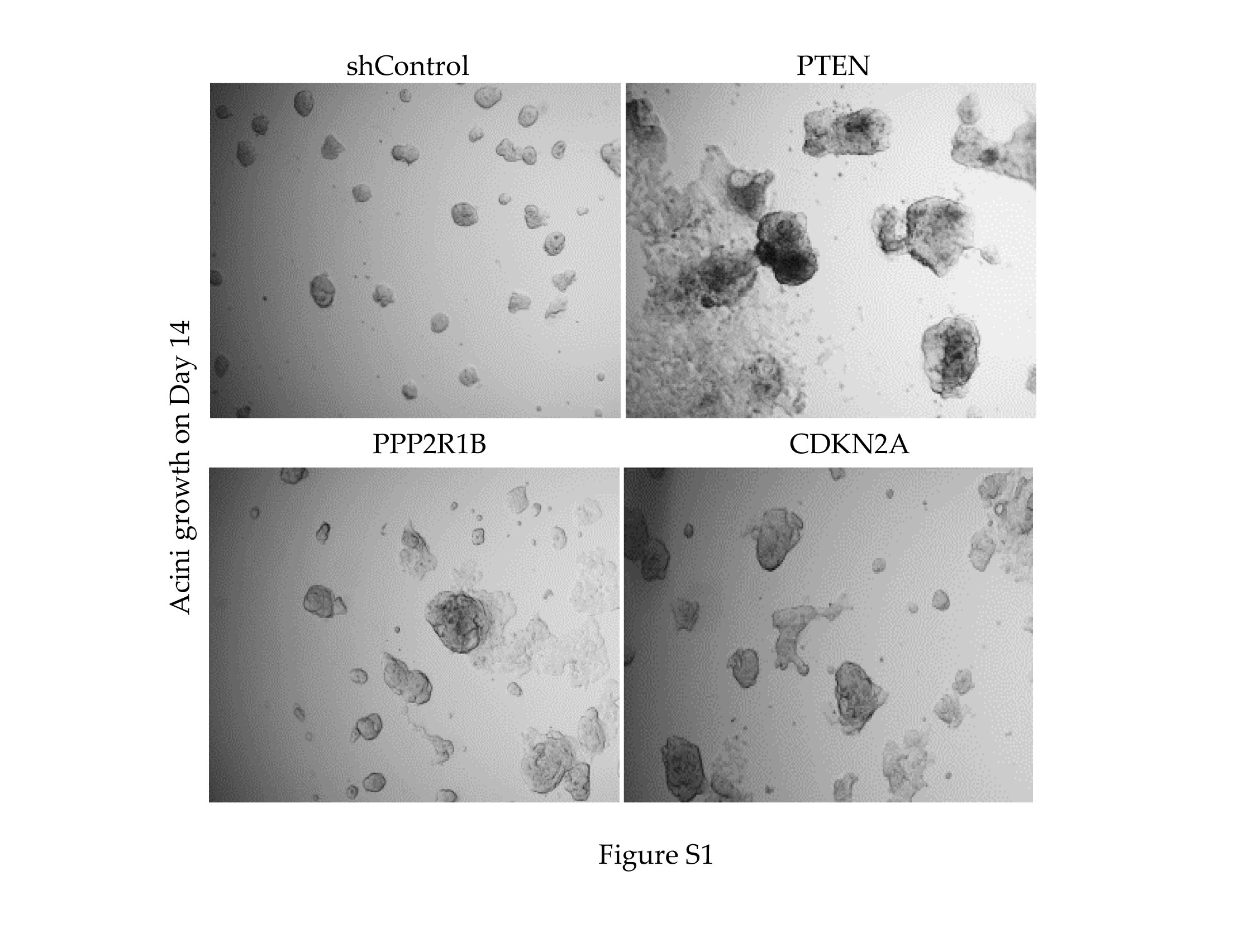
We set out to indentify genes that facilitate spheroid growth from a low proliferative and non-invasive state into larger and more aggressively spreading clusters as would occur when dormant tumour cells resume growth. We adopted the 3D culture model described by Barkan et al28, in which cancer cells of interest are cultured on top of a collagen rich ECM (Matrigel) that mimics the tumour microenvironment more closely than standard cell culture. We tested different serum concentrations to identify a concentration that supported preferential acini outgrowth of invasive basal MDA-MB-231 cells, compared to non-invasive luminal T-47D cells, settling on 2% foetal calf serum (FCS) and growth factor reduced (GFR) Matrigel (data not shown). When cultured in 2% FCS, MDA-MB-231 cells form large colonies with projections, indicative of an invasive phenotype, while T-47D cells remain as small rounded colonies (Fig. 1A). We asked if shRNA-mediated depletion of known tumour suppressors would facilitate aggressive acini formation in T-47D cells. Cells depleted for known tumour suppressor genes, PTEN, PPP2R1B, and CDKN2A formed larger acini, indicating that this approach was able to enrich for cells with enhanced growth potential of T-47D cells in these nutrient poor conditions (Fig. S1A).
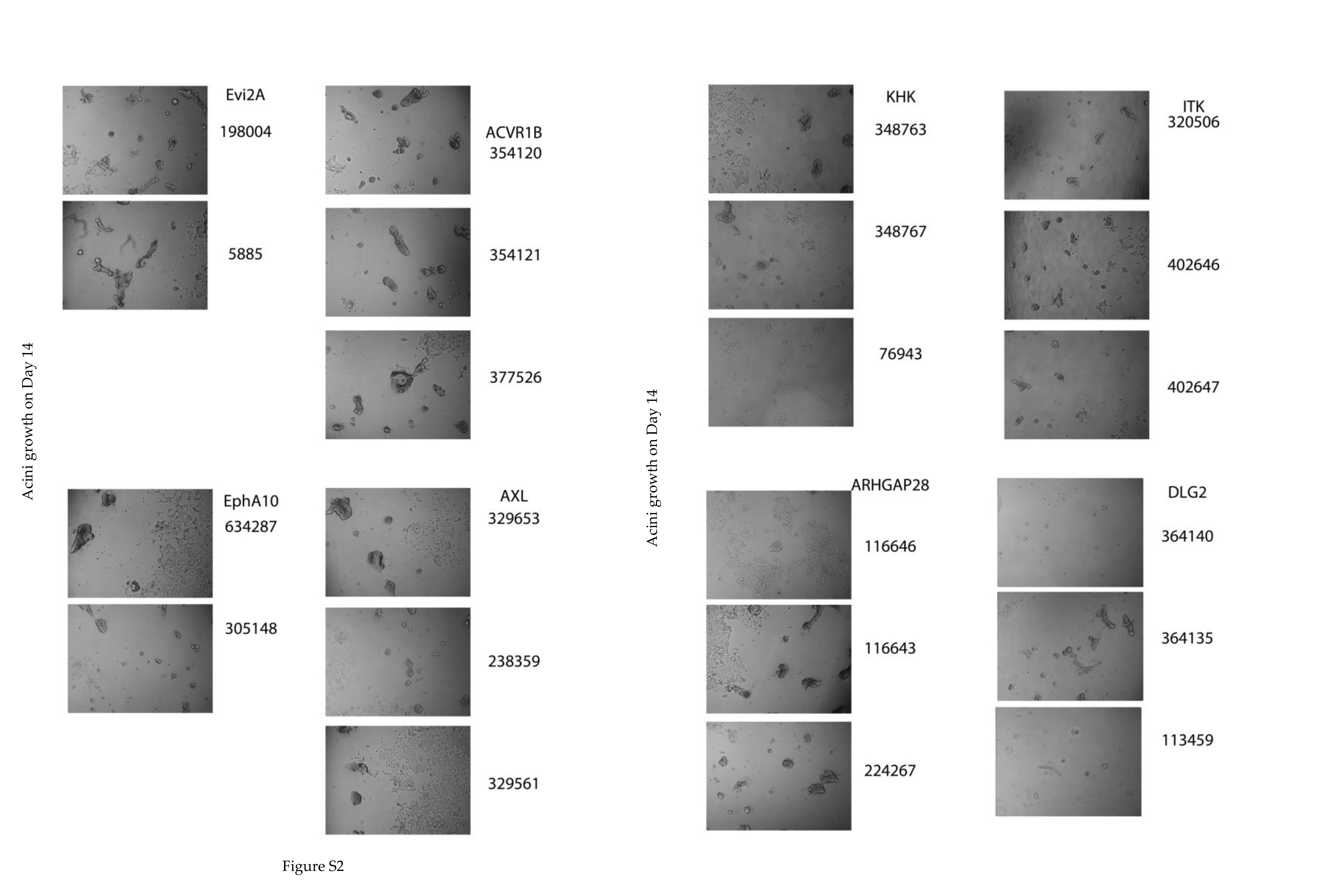
We then completed two screens using the Open Biosystems pGIPz pooled boutique shRNA libraries, one for the kinome and one for polarity genes that were custom designed by the Victorian Centre for Functional Genomics (Peter MacCallum Cancer Centre, Melbourne). T-47D cells were transduced at < 0.5 MOI, maintained at high representation, puromycin selected for 3 days, followed by, culturing on growth factor reduced Matrigel for 14 days. To identify changes in shRNA abundance, genomic DNA isolated from day 0 (starting material) and day 14 acini were used to amplify shRNA sequences that had integrated into the genome using primers targeting the barcoded shRNA sequences as previously reported29,32. Relative plasmid read counts were used to quantify enrichment of shRNAs from day zero to day 14 of 3D culture, and when normalised across the screen, allowed us to calculate the relative enrichment of specific shRNAs on day 14 compared to day zero (Fig. 1B). Genes with two or more hairpins that were > 2-fold enriched on day 14 acini culture compared to day zero were identified (Tabe 1 as Fig. 1C, Table S1), and a selection was validated in a single hairpin per well approach. Specific knockdown of COMMD3 confirmed its potential role as a tumour suppressor (Fig. 2A), while other genes, including several known regulators (AXL, ACVR1B, and DLG2) were also potential suppressors of the invasive phenotype in T47D cells, showing enhanced growth when the gene was knocked down (Fig. 1C and Fig. S2, Table S1).
Commd3 As Novel Tumour Suppressor In Breast Cancer
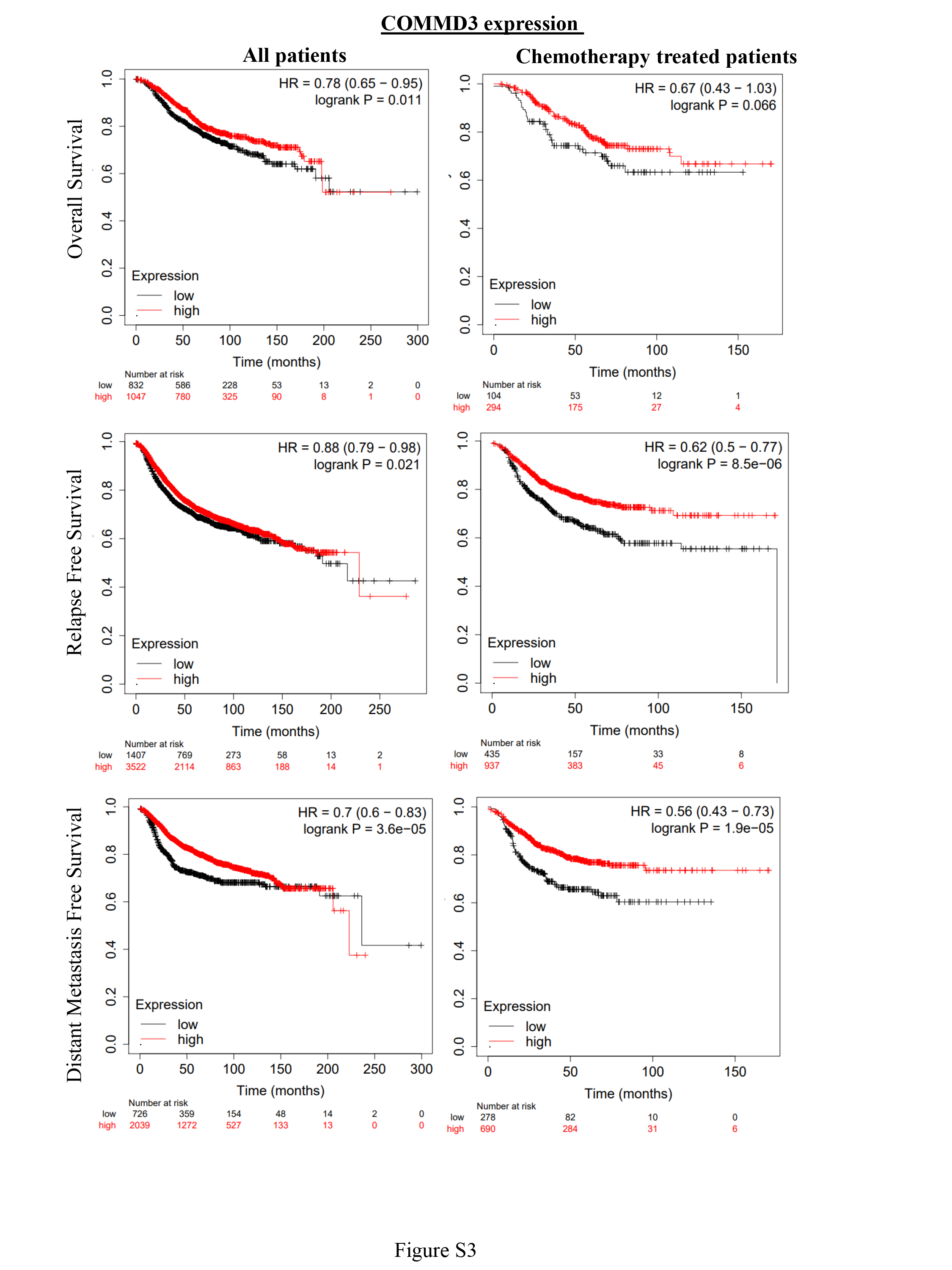
To further investigate novel candidate gene from our screen, we focused our attention on the poorly characterised gene COMMD3. COMMD3 belongs to family of ten proteins (COMMD1-10), characterised by a structurally conserved, C-terminal COMM protein interaction domain, but possessing divergent N-termini33. The canonical family member COMMD1 is a tumour suppressor with cytoplasmic roles controlling vesicular trafficking, and nuclear roles restraining HIF-1α and NFkB activity34,35. We found that shRNA-mediated depletion of COMMD3 resulted in cells with larger acini than the shControl cells (Fig. 2A). This observation was consistent with the notion that COMMD3 loss is responsible for more aggressive and invasive acini outgrowth in breast cancer. We next investigated COMMD3 transcript levels in normal breast and breast tumours by analysing published gene expression datasets. Amongst normal breast epithelial cell subtypes isolated by FACS36, COMMD3 expression was highest in mature luminal epithelia, followed by luminal progenitor and then the basal cell population (myoepithelial + mammary stem cells (stroma)) (Fig. 2B). Consulting the human protein atlas (n = 3 independent normal breast tissue samples stained with a different antibody) confirmed strong expression of COMMD3 protein in terminal ductal alveolar units, polarised toward the lumen37. In the TCGA breast cancer cohort, COMMD3 RNA was highest in luminal and HER2 + tumours, compared to basal- and normal-like subtypes (Fig. 2C). Lower tumour expression of COMMD3 mRNA was associated with a relatively lower probability of survival in the TCGA cohort (Fig. 2D). Analysis of the KMPlotter database showed that these trends were upheld in a range of systemic therapy subgroups (Fig. S3).
To confirm that expression of COMMD3 protein in the tumour cell compartment is associated with disease-specific survival, we performed immunohistochemistry (IHC) analysis of an independent cohort using a commercially available antibody that we first validated on formalin-fixed, paraffin-embedded HEK293T cell pellets (Fig. 2E), showing visibly reduced staining in shCOMMD3 compared to control cultures. A modified COMMD3 IHC protocol was then applied to a consecutive series of invasive breast tumours sampled in tissue microarrays (Table-2). This revealed a range of protein expression levels from negative through to strongly positive, and heterogeneity within individual samples. Staining was captured by an IHC score (intensity x stained area), then distilled into five categories representing increasing levels of overall expression in the tumour compartment (Fig. 2F). Consistent with the mRNA-level analyses, COMMD3 protein expression was significantly higher in ER + and HER2 + tumours compared to TNBCs (Table 3). Interestingly, COMMD3 was inversely associated with the tubule score component of histological grade; that is, tumours that retain some of the polarization and alveolar-like structures typical of normal breast tissue were more likely to express higher levels of COMMD3 (Table 3; ChiSq p = 3.0E− 04).
There were no significant associations with survival in either TNBC or HER2 + groups (not shown), but COMMD3 loss in ER + HER2-negative tumours was associated with poorer survival outcomes, particularly luminal A-like cases (Ki67 staining in ≤ 20% of tumour cells; Fig. 2G). Notably, COMMD3 only stratified survival in this group after about seven years. In other words, for patients with luminal-A-like breast cancer who were still alive seven years after diagnosis, survival probability was lower for tumours lacking COMMD3 (RMST, 10 years). In luminal A-like tumours, COMMD3 was significantly associated with markers of luminal differentiation and/or fate commitment: tubule formation, androgen receptor (AR)38, ELF539 and cKIT36 (Fig-2H; ChiSq p < 0.05).
Loss Of Commd3 Enhances Tumourigenic Potential In Breast Cancer
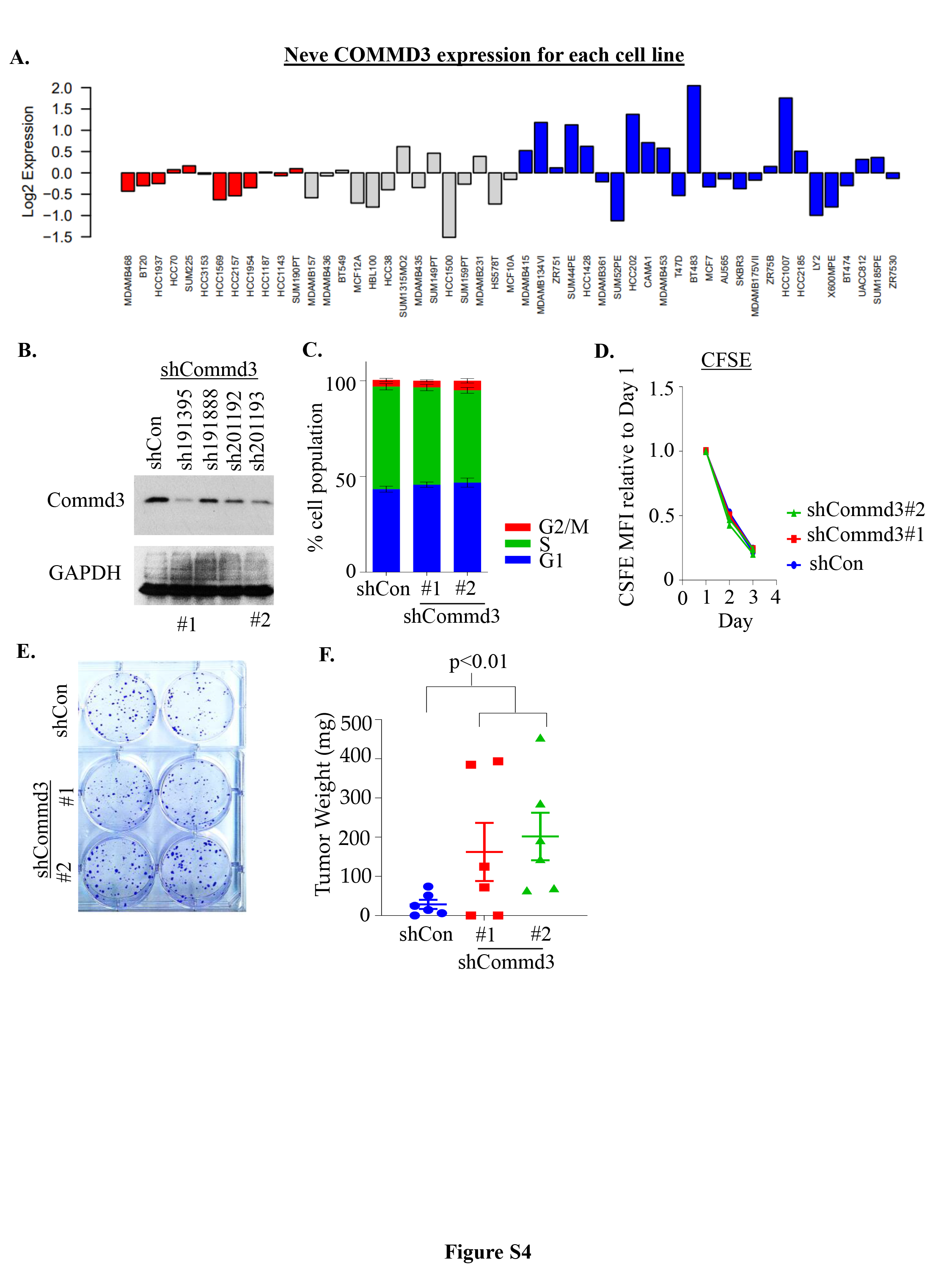
Given the enrichment of shCOMMD3 cells in acini from our genetic screen and the robust expression of COMMD3 in well-differentiated, lower grade breast cancers, we next explored the tumour suppressor function of COMMD3 in more detail. We found that the aggressive and invasive basal-like breast cell lines, particularly the basal B lines exhibited lower COMMD3 expression at both protein and transcript levels compared to luminal cell lines (Fig. 3A-B, Fig. S4A). Moreover, subcellular fractionation of COMMD3 in HEK293T cells demonstrated that COMMD3 is higher in the cytoplasmic insoluble compared to the soluble cytosolic fraction (Fig. 3C). We also found a lower level of COMMD3 in the nuclear fraction, indicating that COMMD3 expression is pan-cellular (Fig. 3C). To study COMMD3 function in breast cancer, we utilised non-metastatic 4T07-TGL (tagged with TK-GFP-luciferase) cells that are syngenic to Balb/c mice. We generated a panel of shCommd3 cell lines and identified one with strong depletion (hairpin 191395#1) and one with partial depletion (hairpin 201193#2) of Commd3 (Fig. 3D, Fig. S4B). We found that knockdown of mCommd3 did not alter proliferation rate, cell cycle distribution, or colony forming capability of cells in 2D culture (Fig. S4C-E). However, it did increase acini growth in 3D culture (Fig. 3E). To explore the role of mCommd3 tumour growth in vivo, a low number of tumor cells (100,000 cells) was engrafted into the mammary fat-pad of Balb/C mice. Interestingly the cell lines with nearly complete or partial depletion of Commd3 had the similar growth characteristics (Fig. 3F and G), indicating Commd3 haplo-insufficiency. Moreover, the shCommd3 cells formed palpable tumours with a shorter latency period and larger tumours at endpoint than their respective shControls tumours (Fig. 3H and Fig. S4F). Collectively, our data provided strong evidence that COMMD3 expression negatively controls breast cancer growth in 3D culture and in vivo..
Whole-transcriptome Profiling Identifies Commd3-regulated Networks In Breast Cancer
COMMD1 has been shown to inhibit NFkB and HIF1α mediated transcription and there is evidence that COMMD3 might do so, albeit to a more limited extent40,41. To gain insight into COMMD3 regulated transcriptional networks, we analysed the transcriptome of 4T07 shControl compared to cells expressing one of two independent shCOMMD3 hairpins in experimental duplicate (two independent samples each of one control and two knockdown hairpins, both with > 90% mCommd3 knockdown). As expected, mCommd3 was the most downregulated gene in our analysis, confirming extensive knockdown (Fig. 4A). Compared to control cells, shCommd3 cells displayed 202 genes with > 1.5-fold downregulation and 43 genes with 1.5-fold upregulation (Fig. 4A, B and Table S2). Consistent with our immunohistochemical analysis of the breast cancer TMA and the shRNA knockdown phenotype, gene set enrichment analysis (GSEA) found that shCommd3 cells were enriched for breast cancer progenitor and pseudopodia haptotaxis signatures (Fig. 4C). Such transcriptional reprogramming could underpin the outgrowth of breast cancer cells with Commd3 loss. We did not observe changes in NFkB and HIF1α mediated transcription as previously reported for COMMD1, suggesting that COMMD3 is part of neither NFkB nor HIF1α networks. Notably, the most highly upregulated gene (2.4-fold, p = 2.74E-06) in shCommd3 cells was the Na+/K + transporter ATP1B1 (Fig. 4A). Upregulation of ATP1B1 transcripts was validated independently, revealing 2–3 fold increase upon Commd3 loss in 4T07 cells (Fig. 4D). ATP1B1 has been reported to be upregulated in response to copper overload. This provides an initial hint that, similar to Commd1, Commd3 deficiency causes changes in copper homeostasis in breast cancer.
Commd3 loss enhances copper overload in breast cancer.
It was interesting to note that ATP1B1 can be upregulated by copper toxicosis. Given the established role of COMMD1 in mediating intracellular copper levels, we hypothesised that COMMD3 deficient cells could also have changes to copper metabolism, involving upregulation of ATP1B1. We therefore assessed intracellular copper abundance by ICP-MS of whole cell lysates. Although not statistically significant, we found that the shCommd3 4T07 cells have slightly increased intracellular copper levels relative to shControl (Fig. 5A). Copper accumulation was assessed in two lines of the murine 4T1 metastasis model (66c14 and 4T1.2) using the Cu+-specific fluorescence-based sensor, Copper Fluor-4 (CF4). The highly metastatic 4T1.2 line in which Commd3 is lowly expressed had significantly higher levels of copper compared to the poorly metastatic 66cl4 cells (Fig. 5B). ATP7A, the major copper-transporting ATPase that regulates copper homeostasis42 was markedly increased in shCommd3 knockdown 4T07 cells compared to shcontrol cells (Fig. 5C), further indicating that Commd3 controls copper homeostasis through ATP7A. To see if depletion of copper would reduce the invasive phenotype of Commd3 deficient cells, we employed the clinically approved copper chelator Tetrathiomolybdate (TM). Treatment of shCommd3 acini with TM significantly reduced the size of acini (Fig. 5D), concomitant with marked induction of apoptosis, as assessed by cleaved PARP (Fig. 5E). However, we did not see any change in ATP7A levels upon TM treatment. These results indicate that copper chelation could be controlling the aggressive and invasive phenotype of COMMD3 deficient breast cancers.
{kind=link}
{kind=link}
{kind=link}
{kind=link}