Cholangiocarcinoma (CCA) and hepatocellular carcinoma (HCC) are deadly malignancy with poor prognosis and limited treatment options. Endoplasmic reticulum (ER) stress plays an important role in the pathogenesis and development of malignant solid tumors which is associated with chemotherapeutic drug resistance. The therapeutic potential of targeting ER stress signaling in cancer via surface BiP/GRP78 (78-kDa glucose-regulated protein), a major role in ER stress sensing, is now under clinical trials. YUM70 is a novel inducer of ER stress that induces apoptosis in cancer by directly bound BiP and inactivated its function. In this study, we investigated the possible role of epidermal growth factor receptor (EGFR) pathway and cell death mechanisms in YUM70 induced CCA or HCC cells cytotoxicity. Although both YUM70 and HA15 as BiP inhibitors exerted the mono-therapeutic anti-proliferation effect and induced autophagy and apoptosis, YUM70 exhibited more potent anti-tumor potential by suppressing the EGFR downstream signaling: ERK1/2 and mTOR/p70(S6K) pathways at the concentration of 100 µM more effectively. At the same tested concentration, HA15 could not inhibit the phosphorylation of ERK1/2 or p70(S6K). Moreover, we discovered that YUM70 induced GSDME dependent pyroptosis by activating NF-κB pathway and inhibited EMT via inactivation of β-catenin pathway. Additionally, pharmacologic targeting of ERK signaling is usually limited by adaptive resistance, frequently mediated by feedback activation of receptor tyrosine kinases (RTKs) signaling. We observed that treatment of HuCCT1 or Huh7 cells with YUM70 resulted in increased EGFR phosphorylation. Inhibiting EGFR activation with Gefitinib or Osimertinib synergistically increased the anti-tumor activity of BiP inhibitors. Our results demonstrated novel strategy that BiP inhibitors, in combination with Gefitinib or Osimertinib, should be tested in CCA or HCC patients.
Research Article
One Novel BiP/GRP78 Inhibitor YUM70 Induces GSDME Dependent Pyroptosis and Enhances Sensitivity To EGFR Inhibitors in cholangiocarcinoma and hepatocellular carcinoma
https://doi.org/10.21203/rs.3.rs-2349164/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
pyroptosis
GSDME
autophagy
apoptosis
ER stress
BiP(GRP78)
EGFR
combination therapy
Hepatocellular carcinoma (HCC), which is a primary liver tumor, is ranked the sixth most common and the fourth most deadly cancer without curative methods[1, 2]. Cholangiocarcinoma (CCA) is the second most common primary liver tumor which is classified anatomically as intrahepatic (iCCA), perihilar (pCCA), or distal CCA (dCCA)[3]. The poor understanding of CCA and HCC pathogenesis hinders the development of effective targeted therapies with their incidence is growing globally. More, CCA or HCC is less sensitive to chemotherapy compared with other solid tumors due to their typical characteristics, such as high levels of fibrosis and profound genetic heterogeneity leading to chemo-resistance[3–6]. The current first-line or second-line targeted therapy mainly contains inhibitors of tyrosine kinases (TKI) and monoclonal antibodies with immune checkpoint inhibitors (ICIs) in HCC or CCA [2, 7, 8]. However, the low response rate of CCA and HCC patients and the acquired resistance to targeted therapies is increasing more and more attention in the world. Additional treatment strategies specifically targeted for CCA and HCC that can overcome the chemo-resistance and improve survival are urgently required.
Increasing evidence show that solid cancer including CCA and HCC can harness endoplasmic reticulum (ER) stress to survive and adopt a drug resistant phenotype through several complex and intertwined resistance mechanisms[9, 10]. The endoplasmic reticulum is the central intracellular organelle which is responsible for synthesizing, folding and modifying secreted and transmembrane proteins to sustain proteostasis[11, 12]. Perturbations in ER function, a process referred to as ‘ER stress’, evoke an adaptive mechanism by triggering the unfolded protein response (UPR), a tightly orchestrated collection of intracellular signal transduction reactions designed to restore protein homeostasis[13]. The UPR sensors contains three primary ER transmembrane signaling receptors named inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like endoplasmic reticulum kinase (PERK), and activated transcription factor 6 (ATF6), which remain inactive when they are bound to ER chaperone BiP/GRP78 (78-kDa glucose-regulated protein), a major role in ER stress sensing[9, 11, 12]. A variety of intrinsic and external stimuli, such as increased protein secretion or disrupted ER protein folding, can lead to the accumulation of misfolded or unfolded proteins in the ER, which leads to dissociation of BiP from these complexes and subsequent activation of UPR in an effort to alleviate cell stress to restore ER homeostasis[9]. In case of extravagated or prolonged conditions, ER stress cannot be reversed and cellular dysfunction deteriorates to cell death which is a condition named ER stress mediated cell death[12, 13]. Thus, enhancement of the UPR activation by designed anti-tumor chemicals represents a promising therapeutic approach for cancers with hyperactive ER stress response that can conquer the ER stress mediated chemoresistance.
ER chaperone BiP/GRP78 is the primary sensor and master regulator of the ER stress response. Elevated levels of the BiP in solid tumor are frequently present in a diverse variety of malignancies, which is associated with increased proliferation rate, invasion and migration potential, chemotherapeutic resistance and poor prognosis in different types of cancer[11, 14–16]. Therefore, small chemicals targeting cell surface BiP appear to be a novel and valuable therapeutic approach that elicits marked anti-tumour effects by disrupting ER homeostasis and inducing cancer cell death[17–21]. For instance, HA15 as one compound of thiazole benzenesulfonamides has been developed to be one anticancer small chemical that could induce ER stress mediated autophagy and apoptosis by targeting BiP in cancer[20, 21]. Recently, the hydroxyquinoline analogue YUM70, which is discovered via a high throughput drug screen, has been confirmed to induce ER stress meditated apoptosis with no toxicity to normal tissues in preclinical models[22]. However, whether YUM70 could function to suppress the growth of CCA or HCC cells has never been investigated. Furthermore, although the anti-tumor effects of BiP inhibitors in some other cancer cells are well established, the cell death mechanisms underlying BiP inhibitors induced cytotoxicity remains elusive especially in CCA and HCC.
Pyroptosis is a newly discovered form of programmed cell death (PCD) triggered by specific Caspases that are mainly activated by inflammation, anticancer drugs, and endoplasmic reticulum (ER) stress[23–26]. Pyroptosis can be mediated by activation of the gasdermin family executor proteins: gasdermin E (GSDME), or gasdermin D (GSDMD), which results in the release of their N-terminal effector domain[23, 27, 28]. Cleaved GSDME or GSDMD fragments then form pores in the cell membranes and trigger perforation, resulting in infiltration of extracellular material and cell swelling followed by pyroptosis[23, 27, 28]. Caspase-3, a well-known apoptotic effector caspase, trigger GSDME-dependent pyroptosis[29]. In the Caspase-3 dependent cell death, GSDME is cleaved and activated by activated caspase-3, which converts non-inflammatory apoptotic death into inflammatory pyroptotic death in cells that express GSDME[23, 27, 30, 31]. The capacity of GSDME to induce cell death determines its critical role in diseases[32]. To date, the functions of GSDME have recently been reported in various solid cancer, which is more strongly implicated in tumor suppression[30, 32]. However, the function and mechanism of GSMDE dependent pyroptosis have not yet been fully clarified. Since GSDME-dependent pyroptosis could suppress tumor growth by activating CD8+ T cell dependent anti-tumor immunity[33, 34] and enhancing chemotherapeutic drug susceptibility[35], induction of GSDME-dependent pyroptosis might be one potential therapeutic approach in solid tumor. Thus, we aimed to explore the role of pyroptosis in BiP inhibitors, including YUM70 and HA15, induced CCA and HCC cell death in this study.
Recent studies demonstrated that epidermal growth factor receptor (EGFR) is overexpressed in CCA and HCC and contributes to development, progression, and chemo-resistance[4, 6, 36, 37]. Mechanistically, EGFR regulates cell proliferation, survival, differentiation and migration mainly through activation of phosphatidylinositol-3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) cascade and mitogen-activated protein kinase (MAPK/ERK1/2) signaling pathway[37, 38]. Therefore, Small molecule tyrosine kinase inhibitors (TKIs) Targeting EGFR is one of the promising therapeutic approaches for CCA and HCC[16, 37]. For instance, EGFR inhibitors showed great efficiency in prolonging survival as monotherapy in patients with lung cancer[39, 40]. However, several independent clinical trials have reported very poor responses in patients with CCA treated with different TKIs and monoclonal antibodies specifically targeting EGFR[39]. Unfortunately, little is known about the factors that limit anti-EGFR responsiveness in CCA. Besides, several recent studies confirmed that feedback activation of EGFR signaling caused by targeted therapeutic drugs could maintain oncogenic signaling and limit cell death in CCA and HCC[4, 41]. Therefore, new approaches to predict and prevent inherent and acquired resistance to EGFR TKIs are urgently needed. Chemotherapeutic resistance typically enables cancer cells to escape the cytotoxicity effect of monotherapy. Combinatorial therapeutic strategies offer a solution for this major challenge. Recently, several combination therapies targeting EGFR pathway have been developed successfully to enhance the chemosensitivity and overcome TKI refractory in HCC and CCA[4, 41, 42]. For example, the combination of the EGFR inhibitor and lenvatinib displays potent anti-proliferative effects in vitro and in vivo in HCC that express EGFR and in vivo in xenografted liver cancer cell lines[4]. Therefore, our study aimed to explore the role of EGFR pathway in YUM70 induced CCA and HCC cell death which could provide one promising targeted therapy by combining YUM70 and EGFR inhibitors.
In the present study, we explored the therapeutic efficacy of YUM70 alone or in combination with EGFR inhibitors. At first, utilizing CCA(HuCCT1 and HCCC9810) and HCC (Huh7) cell lines, we demonstrated that YUM70 inhibited CCA and HCC cancer cell growth more potently than HA15. Base on the finding, we investigated the role of EGFR pathway in YUM70 induced CCA and HCC cytotoxicity, which includes downstream ERK1/2 signaling pathway and mTOR/S6K cascade. To elucidate the molecular mechanism of action underlying YUM70’s direct cytotoxicity to CCA and HCC cells, we examined three different types of programmed cell death including autophagy, apoptosis and pyroptosis in CCA and HCC cell lines. Finally, we discovered that the addition of EGFR inhibitors (1st generation Gefitinib or 3rd generation Osimertinib) abrogates this EGFR reactivation induced by BiP inhibitors and sensitizes cells to YUM70 or HA15 treatment, resulting in a synergistic combination.
Chemicals and reagents
YUM70 were purchased from Selleck (TX, USA) and HA15 from MedChemExpress (NJ, USA). Both BiP inhibitors were dissolved in DMSO to a concentration of 100 µM. Gefitinib in DMSO was purchased from MedChemExpress (NJ, USA) and Osimertinib from Selleck (TX, USA) using as an inhibitor of EGFR. Osimertinib (5 mg) was dissolved in DMSO to make a 10 mM stock solution.
Cell culture
Human cancer cell lines including HuCCT1, Huh7 and HepG2 cells were obtained from National of Authenticated Cell Cultures (Shanghai, China) and HCCC9810 cells was kindly granted by Professor Haojie Jin (Shanghai Jiaotong University, Shanghai, China). The cells were grown in Dulbecco's modified Eagle's medium (DMEM, Gibco, CA, USA) or Roswell Park Memorial Institute (RPMI) 1640 (RPMI 1640, Gibco, CA, USA) containing 10% fetal bovine serum (FBS, Vivacell Bioscience, Shanghai, China) and 100 U/ml penicillin-streptomycin (Gibco, CA, USA) in a humidified incubator of 5༅ CO2 at 37˚C.
Small interfering RNA (siRNA) transfection
siRNA targeting BiP (5‘-CUUAAGUCUCGAAUGUAAUTT-3’; 5‘-GGUGGGCAAACAAAGACAUTT-3’) and negative control siRNA (5’-UUCUCCGAACGUGUCACGUTT-3’), which was used for normalization, were synthesized by Genepharma (Shang Hai, China). For transfection, Huh7 or HCCC9810 cells were seeded in 6-well plates at a density of 2x105 cells/well. After 24 hours, cells were transfected with siRNA (100 pmol) using Lipofectamine® 2000 Reagent (Invitrogen, CA, USA) according to the manufacturer’ instructions. After 48 hours of transfection, HCCC9810 cells were treated with 100 µM YUM70 for another 24 hours. Then the cells were harvested and knockdown of BiP was confirmed by western blot analysis.
Western blot analysis
Monolayer cultures of respective cell lines at an 80%-90༅ confluence were prepared with pre-cold Ripa lysis and extraction buffer (Sangon Biotech, Shanghai, China) containing protease and phosphatase Inhibitor Cocktail (Thermal Scientific, CA, USA) on ice. The total cell lysate was centrifuged and the supernatant was denatured by boiling. Protein concentrations of supernatants were analyzed by bicinchoninic acid (BCA) assay kit (Beyotime, Nantong, China). Equivalent amounts of total proteins (20µg) were subjected to 6༅-12༅ sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a 0.22 µM PVDF membrane (Millipore, MD, USA). The membranes were blocked for 2 h in 5༅ bovine serum albumin (BSA) at room temperature and incubated with specific primary antibodies at 4˚C overnight. A list of the primary antibodies used for Western blot are characterized in Table 1. Further incubation was performed with the corresponding horseradish peroxidase-coupled secondary antibodies (1:2000, Cat. sc-516102; Santa Cruze, CA, USA & 1:3000, Cat. #7074; cell Signaling technology, Inc. (CST), MA, USA) at room temperature for 2 h. Then the bands were detected using Super Signal® West Pico Chemiluminescent Substrate kit (Thermo Scientific, CA, USA), and the results were recorded using the ChemiDoxTM XRS + system. Relative protein expression was normalized with β-actin.
Table 1
Primary antibodies used in western blot analysis.

Crystal Violet Staining
Huh7 or HCCC9810 Cells were seeded in an equal number per well (0.3 × 105 cells/well) on 12-well plates and allowed to attach overnight. Fresh media with or without indicated drugs were replaced the next day. After treatment for 48 hours, cells were fixed in 4%paraformaldehyde solution for 10 minutes and subsequently stained with 0.5༅ crystal violet solution for 1 minute. Plates were washed three times with PBS and dried overnight.
Colony formation assay
HuCCT1, HCCC9810 or Huh7 cells (300 to 500 cells/well) were grown in 6-well plates for 2 days. Cells were treated with BiP inhibitors and EGFR TKIs alone or combination for 24 hours or 48 hours. Fresh media without drugs were replaced and refreshed every 2–3 days until the end of the experiments. Following methanol fixation, colonies were stained with 0.5% crystal violet. Plates were washed three times with PBS and dried overnight.
Statistical analysis
All data from the colony formation assay are presented as means ± SD of at least three experimental replicates. To compare differences between YUM70 group and HA15 group at the same concentration (50, 100, 200 µM), one-way ANOVA test was performed and values of P ≤ 0.05 were considered statistically significant.
YUM70 induces ER stress in CAA and HCC cell lines
At the first step, we evaluated whether the novel BiP inhibitor YUM70 could activate the UPR signaling pathways, which are ER stress markers, in CCA and HCC cells. HA15 is another BiP inhibitor compound that exhibits strong efficacy in anti-tumor activity by targeting BiP and triggering unresolved ER stress[20]. Thus, we examined the ER stress induced by YUM70 or HA15 by using Western Blot. BiP, phospho-eIF2α and CHOP as Key ER stress sensors were increased and total ATF6 was decreased by YUM70 or HA15 in HuCCT1 or HepG2 cells respectively (Fig 1A and B). And we compared the ability of ER stress induction of YUM70 and HA15 in HuCCT1 cells by detecting BiP, inositol-requiring enzyme 1α (IRE1α), phospho-JNK, and phospho-eIF2α (Fig. 1C). Similar JNK pathway activation was also verified in YUM70 or HA15 treated Huh7 cells (Fig. 1D). These results demonstrated that YUM70 and HA15 are both BiP inhibitors with similar efficiency and mechanisms involved in ER Stress induction in CCA and HCC.
YUM70 impairs the colony forming ability of CAA and HCC cell lines
At the second step, we evaluated the therapeutic potential of both BiP inhibitors for the treatment of CCA or HCC. The cell growth inhibitory effects of YUM70 or HA15 in CAA and HCC cells were determined using the colony formation assay. Briefly, CCA cell lines (HuCCT1 and HCCC9810) and HCC cell line (Huh7) were treated with YUM70 or HA15 at concentrations of 50, 100, 200 μM or vehicle control (DMSO) for 24 hours followed by fresh drug-free media culture for 10-14 days. As shown in Fig. 2A, whether YUM70 or HA15 significantly reduced the colony-forming ability of CAA and HCC cells in a dose-dependent manner. Interestingly, Colony formation experiments also revealed that HuCCT1 and HCCC9810 cholangiocarcinoma cells and Huh7 hepatic cancer cells exhibited differential sensitivity to both BiP inhibitors, which could induce ER stress similarly. CCA and HCC cells under HA15 treatment displayed a better survival than those under YUM70 treatment with an equal amount (Fig. 2A). It was identified that CAA and HCC cells were more sensitive to YUM70 than HA15 at the same concentration (50, 100 or 200 μM) respectively (Fig. 2B-D).
YUM70 is more potent in inhibiting ERK1/2 and p70(S6K) pathways than HA15.
Considering that an equal amount of YUM70 or HA15 resulted in different anti-tumor activity in CCA and HCC cells (Fig. 2), we investigated whether the differences in inhibiting activity of EGFR pathway, which is one fundamental role in CCA and HCC, produced the differential result between the YUM70 and HA15. Treatment of HuCCT1 and Huh7 with YUM70 resulted in time or dose dependent inhibition of phosphorylation of ERK and p70(S6K), while phosphorylation levels of both proteins remained unchanged or elevated in response to HA15 (Fig. 3A-C). And in HCCC9810 CCA cells, YUM70 also induced ER stress which was verified by the increased expression of BiP, IRE1α, phosphor-JNK and PERK and the decreased expression of total ATF6 (Fig. 3D). In HCCC9810 cells under 100 μM YUM70 for 24 hours, phosphor-EGFR, phosphor-ERK, phosphor mTOR and phosphor-S6K proteins were significantly decreased concomitantly with ER stress (Fig. 3D). Thus, YUM70 exhibited strong inhibitory activity on EGFR/ERK1/2 and mTOR/S6K pathways. These data showed that YUM70, not HA15, at the tested conditions of 100 μM at 24 hours effectively decreased the levels of p-ERK and p-p70(S6K), two well-known events downstream of EGFR, in CAA and HCC cell lines (Fig 3A and B).
As well known, EGFR downstream ERK and S6K pathways play the vital roles in sustaining cell proliferation and preventing cells death in CCA and HCC[7, 43-45]. These data indicated that YUM70 suppressed CAA and HCC cells proliferation more potently than HA15 under the tested conditions via inhibiting ERK and S6K pathways more efficiently (Fig. 2 and 3A and B).
YUM70 induced autophagy, apoptosis and GSDME dependent pyroptosis.
ER stress could induce autophagy and apoptosis by induction of the pro-apoptotic transcriptional factor CHOP or activating c-Jun amino terminal kinase (JNK) signaling cascade[13, 46-51]. Since we confirmed that YUM70 treatment activated ER stress and suppress the cell proliferation by inhibiting ERK1/2 and S6K pathways in CCA and HCC cells (Fig. 1-3), we speculated that YUM70 might also activate ER stress-induced autophagy and apoptosis via inhibiting mTOR/S6K and EGFR/ERK pathways in CCA and HCC cells.
Thus, we evaluated the cell death mechanisms induced by BiP inhibitors in CCA or HCC. In HuCCT1, HepG2, HCCC9810 or Huh7 cells, Westen Blot analysis demonstrated a dose or time dependent increase in the cleaved poly (ADP-ribose) polymerase (PARP) fraction and the conversion of LC3B-Ⅰ to LC3-Ⅱ associated with autophagosome formation after YUM70 or HA15 treatment (Fig. 4A-D and S1A). Meanwhile, YUM70 or HA15 decreased p62 or increased ATG5, all of which also serve as the hallmarks of autophagy (Fig. 4A, B and D). These results indicated that both YUM70 and HA15 induced autophagy and apoptosis in CCA and HCC cells as predicted. Furthermore, it has been confirmed that inhibition of apoptosis or autophagy partially inhibited the cell death induced by HA15, whereas concomitant inhibition of both completely restored cancer cell viability[20]. It could be concluded that the mechanisms involved in YUM70 or HA15 induced cell death consisted of autophagy or apoptosis via inhibition of S6K pathway or ERK1/2 pathway in CCA and HCC cells respectively.
Apoptotic cell death can be classified into caspase-3 dependent and caspase-3 independent apoptosis [47]. Therefore, expression of cleaved PARP and cleaved Caspase-3, as evidence of cell death, was measured by Western Blot in HuCCT1 at 12 and 24 hours under the treatment of YUM70 or HA15 at the concentration of 100 μM (Fig. 4C). Both YUM70 and HA15 could increase cleaved PARP fraction and only YUM70 increased cleaved Caspase-3, which could cleave PARP during apoptosis. However, no significant cleaved Caspase-3 band was observed under HA15 treated HuCCT1 cells in line with Fig. 4B in HepG2 cells, thus suggesting that YUM70 induced a canonical Caspase-3 dependent apoptosis and HA15 induced a non-canonical Caspase-3 independent apoptosis.
Since the ER stress might also cause pyroptotic cell death, we examined whether BiP inhibitors induced pyroptosis in CCA and HCC cells. During pyroptosis, the N-terminal domain dimer of GSDME is generated through specific cleavage by activated caspase-3 and induces cell membrane perforation [27, 29, 30, 52-55]. To investigate whether BiP inhibitors could induce pyroptotic cell death, we monitored the cleavage of the pyroptosis effector GSDME. YUM70 treatment induced the cleavage of GSDME in a concentration and time dependent manner in HuCCT1 and HCCC9810 cells (Fig. 4A, C and D). Consistent with the generation of cleaved GSDME, activated Caspase-3 was detected in response to YUM70 alone (Fig. 4C and D). Of note, HA15, comparing to YUM70, could not activate Caspase-3 or produce the cleavage of GSDME (Fig. 4B and C). These results indicated that YUM70 induced GSDME cleavage was related to Caspase-3 activation. In brief, these results indicate that YUM70 at lest induced two types of programmed cell death in CAA and HCC cells: autophagy related apoptosis and caspase-3 mediated GSDME dependent pyroptosis. While, HA15 only induced Caspase-3 independent apoptosis. In summary, the above results indicated that YUM70 not only suppressed cell proliferation but also induced Caspase-3 dependent apoptosis and GSDME–dependent pyroptosis (Fig. 2 and 4).
GSDME dependent pyroptosis is activated by the NF-κb signaling in CAA and HCC cells.
Subsequently, to determine the role of ER stress in YUM70 induced autophagy, apoptosis and GSDME dependent pyroptosis, BiP as the key ER stress sensor was knocked down by siRNA. siRNA Knockdown of BiP could decrease cleaved PARP and LC3Ⅱ induced by YUM70 in HuCCT1 cells (Fig 5A). The results showed that BiP knockdown resulted in a significant resistance to apoptosis and marked decrease of autophagy under ER stress induced by YUM70. However, cleaved GSDME was increased in BiP-knockdown HuCCT1 under YUM70 treatment (Fig. 5A). It indicated that YUM70 triggered pyroptosis, which could not be alleviated by BiP knockdown, was not associated with ER stress induced by YUM70 itself. In addition, ER stress induced by HA15, as another BiP inhibitor, could not induce pyroptosis (Fig. 4B and C). Small molecule inhibitors often have various off-target effect implicated in targeted therapy and resistance. Considering that non-canonical ER stress independent functions, such as Bexarotene triggered pyroptosis with no association with activated UPR signaling, have been identified in several studies [12, 16, 26, 56], our results indicated that YUM70 induced ER stress independent pyroptosis and ER stress dependent autophagy and apoptosis simultaneously.
Next, we aimed to explore the underlying mechanisms of YUM70 triggered pyroptosis in CCA and HCC cells. Several studies have reported that GSDME dependent pyroptosis was found to be regulated by NF-κB or JNK pathway, which also induce apoptosis [47, 54, 55]. Our results showed that both YUM70 and HA15 could activate JNK pathway in CCA and HCC cells (Fig. 1C and D). But HA15 could not activate Caspase-3 or trigger pyroptosis. Thus, it could be excluded that JNK pathway play the key role in YUM70 induced Caspase-3 dependent pyroptosis. Interestingly, Western blot analysis showed that YUM70 increased the phosphorylation of NF-κB in a dose dependent manner in Huh7 and HuCCT1 cells (Fig. 5B and E). In YUM70 treated HuCCT1 cells, phosphorylation of NF-κB was also increased significantly (Fig. 5D). However, HA15 could not increase the phosphorylation of NF-κB in HuCCT1 and Huh7 cells (Fig. 5B and C). HA15 even decreased phosphorylation of NF-κB in a dose dependent manner in HepG2 cells (Fig. 5E). These results indicated that only YUM70 could activate NF-κB pathway which could increase the expression of cleaved Caspase-3, the essential executor protein in GSDME cleavage [54, 57]. Since HA15 could inhibit NF-κB pathway without triggering pyroptosis (Fig. 4C and 5E), it was feasible that YUM70 induced Caspase-3 dependent pyroptosis via NF-κB signaling cascade that HA15 could not activate.
YUM70 suppressed EMT via inhibitingβ-catenin pathway.
Epithelial–mesenchymal transition (EMT) has been confirmed to play a fundamental role in promoting metastasis and invasion of cancer cells[58, 59], which led us to examine the expression of EMT markers, including E-cadherin, N-cadherin, and Vimentin. Immunoblotting analysis showed that the protein expression of epithelial marker E-cadherin was increased, while the expression of mesenchymal markers N-cadherin and Vimentin were markedly reduced in a dose-dependent manner in YUM70 treated HuCCT1 cells (Fig. 6A). The canonical Wnt/β-catenin cascade is one crucial signaling pathway that can driving carcinogenesis and EMT transition[59, 60]. To explore the molecular mechanism involved in suppressing EMT by YUM70, we examined β-catenin expression in YUM70 treated HuCCT1 cells by using Western Blot. β-catenin expression level was also decreased in a dose-dependent manner in HuCCT1 under the treatment of YUM70 (Fig. 6A). HCCC9810 under YUM70 treatment also show decreased expression of mesenchymal markers (N-cadherin and Vimentin) and β-catenin monitored by Western Blot (Fig. 6B). Together, these results suggest that YUM70 suppresses EMT that could promote the migration and invasion potential of CCA cells by inhibiting β-catenin pathway.
BiP inhibitors-based combination treatment in CAA and HCC.
In Figure 3, we observed durably suppression of p-ERK in one time or dose dependent manner in HuCCT1, HCCC9810 or Huh7 cells treated with the YUM70. However, we also observed increased phosphorylation of EGFR at Tyr1068 positions in HuCCT1 or Huh7 cells under treatment with 100 μM YUM70 or HA15 (Fig. 7A). And we observed increased phosphorylation of EGFR without overexpression of total EGFR in one dose dependent manner in YUM70 treated HuCCT1 cells (Fig. 7B). These results showed that the feedback activation of EGFR at phosphorylation site Tyr1068 was significantly increased in response to YUM70, which could durably suppress EGFR downstream MEK1/2/ERK1/2 and mTOR/S6K pathways (Fig. 3).
Our results were consistent with the previous reports that adaptive signaling responses to MEK/ERK pathway inhibition might involve upregulation of upstream signals, including increased responsiveness of RTK[61, 62]. Amplification of the upstream oncogenic driver of ERK signaling has been identified as a mechanism for MEK/ERK inhibitor resistance[61, 62]. On the other side, cancer cells with constitutive or acquired resistance to chemotherapy are also resistant to ER stress mediated cell death [63, 64]. Therefore, these results also suggested a possible role for EGFR in mediating RTK driven adaptive resistance to ER stress mediated cell death induced by BiP inhibitors in these cancer cells. For instance, recent research demonstrated that feedback activation of EGFR limits the response to Lenvatinib in HCC [4].
Previous studies have shown that ERK is reactivated in cancer cells under treatment of EGFR inhibitors[65] and EGFR-MEK dual inhibition significantly delays their emergence[42, 66]. Considering that YUM70 could effectively inhibit downstream ERK1/2 pathway and reactivate upstream EGFR, we developed the combination therapy of YUM70 plus EGFR inhibitors. A large number of studies have confirmed that whether the first generation Gefitinib or the third generation Osimertinib as EGFR inhibitors could inhibit the phosphorylation of EGFR potently in cancer cells [67]. As expected, the combination of Gefitinib or Osimertinib and YUM70 was remarkably effective in inhibiting cell growth in Huh7 or HCCC9810 cell line (Supplement 1C and D). Although HA15 at 100 μM concentration did not inhibit ERK pathway as YUM70 did, HA15 could inhibit ERK pathway at the higher concentration of 200 μM in HepG2 cells (Date not shown). Considering that Osimertinib is more potent in inhibiting EGFR phosphorylation than Gefitinib[67], we next examined the effect of combinations of Osimertinib with YUM70 or HA15 in HuCCT1 cells. For this purpose, we performed colony formation analyses in the presence of Osimertinib and BiP inhibitors (YUM70 or HA15) alone or in combination respectively. Our results showed that the combination of YUM70 (or HA15) and Osimertinib led to a robust growth inhibition of cultured colonies in HuCCT1 cells (Fig. 7C).
Although treatment with YUM70 suppressed ERK1/2 activation without EGFR rebound activation in HCCC9810 cell lines, the combination of YUM70 and Osimertinib also significantly decreased HCCC9810 cell colonies growth (Fig. 7D and E). Overall, HuCCT1, Huh7 and HCCC9810 cell lines showed exquisite sensitivity to combinatorial treatment with YUM70 or HA15 and EGFR inhibitors (Fig. 7C and E and Supplement 1C and D). These results demonstrate that combining these two drugs achieved a synergistic effect regardless of phosphorylation status of EGFR.
In this study, we firstly showed that activated ER stress under the treatment of YUM70 or HA15 with similar efficiency in HCC and CAA cells (Fig. 1), which was consistent with the findings of previous reports wherein ER stress was induced in the human pancreatic and melanoma cell lines [20, 22]. On the basis, this study revealed that YUM70 suppressed CCA and HCC cells proliferation more potently than HA15 via inhibiting ERK1/2 and S6K pathways at the tested concentration of 100 µM (Figs. 2 and 3A and B). Although YUM70 has been reported to induce cell death in pancreatic cancer cell lines[22], the mechanisms of its action are not clear. In this study, we demonstrated that YUM70 induces Caspase-3 dependent cleavage of GSDME via NF-κb pathway and autophagy related apoptosis via ERK, mTOR and JNK pathways (Fig. 1, 3–5). In brief, this study clarified that YUM70 induced cell death might consisted of ER stress dependent apoptosis and GSDME dependent pyroptosis mediated by Caspase-3 activation (Fig. 4C and D). YUM70 triggered pyroptosis was not associated with ER stress induced by YUM70 and via NF-κB pathway which was specifically activated by YUM70, not HA15 (Fig. 5). In addition, YUM70 suppresses EMT that could promote the migration and invasion potential of CCA cells by inhibiting β-catenin pathway (Fig. 6). To the best of our knowledge, this is the first study showing a therapeutic anti-tumor effect of YUM70 monotherapy in CCA and HCC. Finally, we demonstrated that BiP inhibitors reactivated EGFR phosphorylation in Huh7 and HuCCT1 cells but inhibited EGFR/MEK1/2/ERK1/2 cascade in HCCC810 cells (Fig. 7A, B and D). Combining BiP inhibitors and EGFR inhibitors in EGFR reactivated HuCCT1 and Huh7 or EGFR non-reactivated HCCC9810l achieved a synergistic effect regardless of EGFR status (Fig. 7C and E and S1C and D).
YUM70 is one novel hydroxyquinoline analogue with significant efficacy in pancreatic cancer that has been synthesized and screened recently[22]. However, the therapeutic effect of YUM70 in CCA and HCC and its underlying mechanisms has not yet been reported. Initially, our results showed YUM70 activated all of three UPR signal pathways, including PERK, ATF6 and IRE1-α pathways, which was similar to HA15 induced ER stress (Fig. 1). Interestingly, YUM70 suppressed the clonogenicity of CAA and HCC cells more potently than HA15 (Fig. 2). Mechanically, at the molecular level, YUM70 effectively suppressed p-ERK and p-S6K levels and increased p-JNK level in CCA and HCC. In contrast, HA15, which only increased p-JNK lever, could not inhibit p-ERK or p-S6K at the tested condition of 100 µM (Figs. 1 and 3). ERK1/2 activity, which is commonly upregulated in cancer, can serve as a hallmark of various refractory cancers such as HCC and CCA[45]. The phosphorylation activates ERK1/2 and drives its translocation into the nucleus. In the process, ERK1/2 phosphorylates numerous cytoplasmic and nuclear signaling protein substrates such as ribosomal S6 kinase (S6K), which promote cancer cell proliferation, survival, differentiation and chemoresistance[45, 68, 69]. Therefore, highly selective and potent small molecule inhibitors targeting ERK1/2 cascade can exerts anti-apoptotic functions[45, 70]. Meanwhile, blocking ERK1/2 pathway or mTOR/S6K cascade could enhance sensitivity of cancer cells to chemotherapy drugs[4, 69–73]. And numerous studies have confirmed that activation of JNK cascade contributes to cellular stress, such as ER stress, induced apoptosis[47]. Therefore, our results indicate that inhibition of ERK1/2/S6K pathway and ER stress activated JNK pathway might be involved in YUM70 induced antitumor activity against CCA and HCC synchronously.
Subsequently, we evaluated the form of cell death caused by YUM70. Initially, by using western blotting, we determined that YUM70 induced CCA and HCC cells apoptosis by increasing the proportion of the cleavage subtype in key apoptosis markers including poly-PARP and Caspase-3. In addition, YUM70 also increased the conversion of LC3 Ⅰ to LC3 Ⅱ and decreasing the level of p62, both of which are key autophagy markers (Fig. 4A and D). ER stress is frequently accompanied by autophagy via the stimulation of AMPK, which leads to inhibition of the AKT/mTOR/S6K pathway[74]. In the present study, we provided the first evidence that YUM70 induced ER stress mediated autophagy and apoptosis by inhibiting EGFR downstream ERK1/2 and S6K pathways and activating JNK pathways (Figs. 1, 3 and 4).
Both autophagy and apoptosis are synchronously responsible for ER stress mediated cell death triggered by BiP inhibitors[20]. However, whether pyroptosis is involved in the mechanisms of ER stress induced cell death has not yet been reported. For the first time, we uncovered the involvement of YUM70 triggered non-apoptotic form of death, pyroptosis. We confirmed that YUM70 induced pyroptosis is mediated by Caspase-3 activation and GSDME cleavage (Fig. 4). Interesting, HA15, as another BiP inhibitor, could activate PARP but not activate Caspase-3 that can activate GSDME (Fig. 4C). Therefore, Caspase-3 activation and GSMDE cleavage were essential in YUM70 induced pyroptosis. GSDME is a critical effector of pyroptosis which disrupts cell membranes upon cleavage by Caspase-3 in cells expressing GSDME and undergoing apoptosis[23, 29]. By comparing analysis of the cell death mechanisms induced by both BiP inhibitors (YUM70 and HA15), our finding further prompted Shao group’s concept that GSDME could switch chemotherapy drugs induced and Caspase-3-mediated apoptosis to pyroptosis[23]. Caspase activation plays pivotal roles in mitochondria mediated apoptotic pathway. Activates Caspase-3 results in GSDME cleavage (pyroptosis) converting non-inflammatory apoptotic death into inflammatory pyroptotic death[23, 29, 30, 32]. Thus, Caspase-3/GSDME axis is essential for YUM70 triggered pyroptosis in CCA and HCC. Unlike necrosis or apoptosis, pyroptosis occurs in a specific and distinguished procedure in which cell membrane integrity is disrupted to trigger an inflammatory cell death whereby cellular contents, including inflammatory cytokines, are released into the extracellular space[30]. In most studies, pyroptosis could increase chemotherapeutic effectiveness in suppressing tumor growth by activating anti-tumor immunity or enhancing chemotherapeutic sensitivity[30, 33–35]. However, in certain cases, pyroptosis was considered as strong side effects of anti-tumor chemotherapy which could confer resistance to chemotherapeutic toxicity in various solid tumor[29]. Thus, further study is required to determine whether YUM70 induced GSDME dependent pyroptosis play a protective or detrimental role for CCA and HCC. Furthermore, it is necessary to investigate underlying mechanisms for the immune modulation ability of YUM70 triggered pyroptosis in our further study.
BiP was the most important upstream molecular that was involved in YUM70 or HA15 induced ER stress. Our results showed BiP knockdown decreased YUM70 induced autophagy and apoptosis and increased GSDME dependent pyroptosis (Fig. 5A), suggesting that YUM70 induced ER stress dependent autophagy related apoptosis and ER stress independent pyroptosis. One similar study has been reported that GSDME dependent pyroptosis can be independent of bexarotene induced ER stress in ovarian cancer cells because the attenuation of ER stress by siRNA against PERK or IRE1α could not restore the cleavage of GSDME[26]. Indeed, BiP knockdown exacerbated YUM70 induced pyroptosis (Fig. 5A), which indicated that ER stress could protect cancer cells from proptosis. Since ER stress initially serves as a series of compensatory protective responses to protect cancer cells from irreversible damage[11, 47], it is possible that ER stress can also protect cells by suppressing pyroptosis triggered by YUM70. Additional studies are required to address this critical issue about the protective response of ER to pyroptosis.
Besides, EMT is essential for tumors acquiring aggressive features, which mainly consists of metastatic and invasive properties by promoting core procedures of the metastasis and invasiveness signaling cascades[75]. As well, EMT contributes critically to the development of resistance to various types of therapeutic anti-tumor drugs[76]. Indeed, we also demonstrated that YUM70 suppressed the EMT in CCA and HCC cells in a dose-dependent manner (Fig. 6A). Accordingly, YUM70 increases the EMT-related proteins such as E-cadherin expression, while decreased the expression of N-cadherin and Vimentin by inhibiting β-catenin pathway, which could suppress cell migration and invasion ability of CCA and HCC cells.
Collectively, our study revealed that YUM70 triggers programmed cell death (PCD) not via a single specific pathway but rather via multiple pathways that may overlap with each other. Apoptosis, autophagy and pyroptosis are the primary cell death involved in YUM70 induced chemotherapeutic effect in CCA and HCC. In short, Caspase3-dependent apoptosis pathway induced YUM70 triggered GSDME dependent pyroptosis. In YUM70-treated cells, we showed the occurrence of ER stress independent pyroptosis in parallel to ER stress dependent autophagy and apoptosis which are consequences of over-expression of BiP followed by induction of UPR. These findings indicate that YUM70 has a strong anti-tumor effect via modulating multiple malignancy behaviors in vitro.
Furthermore, our study demonstrated that ERK1/2 and mTOR/S6K pathways downstream of EGFR signaling cascade was inhibited by YUM70 without any rebound in CAA and HCC (Fig. 3). Meanwhile, EGFR was selectively reactivated by YUM70 in Huh7 and HuCCT1 cells not in HCCC9810 cells (Fig. 7). Therefore, our results were consistent with one previous study that reactivation of EGFR, as the required upstream receptor of ERK1/2, is not sufficient for ERK1/2 rebound [62]. In addition, the detailed mechanisms of reactivating EGFR by YUM70 in Huh7 and HuCCT1 cells remains elusive in our current study. It has been reported that increased phosphorylation of JNK could enhance the activation of EGFR [77]. Since YUM70 activated JNK pathway downstream of ER stress without reactivating EGFR in HCCC9810 cells (Fig. 3D and 7D), it could be excluded that JNK pathway play the key role in this RTK signaling response. Further study is necessary to fully characterize the mechanism of selective EGFR reactivation in CAA and HCC cells and determine what signaling may be responsible.
Monotherapy often causes adaptive drug resistance which is a major challenge to the clinical success of cancer therapies[78]. In the process, inhibition of the specific target by small molecule drugs might relieve feedback inhibition and resultantly reactivate signaling cascades, which subsequently attenuate anti-tumor effects of targeted therapies[69, 79]. Currently, one clinical strategy that can overcome the acquired resistance is the development of combination therapies based on biological context[78, 80]. Appropriate combination treatment could enhance the therapeutic potential of chemotherapeutic drugs and reduce their toxicity by allowing the administration at a lower dose[73, 78]. For instance, several studies have confirmed that ERK1/2 inhibitor in combination with EGFR TKI could overcome the acquired chemoresistance to EGFR TKIs with favorable tolerability[66, 78]. In the present investigation, we demonstrated that BiP inhibitors synergizes with EGFR inhibitors to suppress CAA and HCC cells growth, supporting the further testing of these combination therapies for the treatment of CAA and HCC. The selection of EGFR inhibitors was based on the pathway analysis using BiP inhibitors monotherapy: YUM70 or HA15 feedback increased the phosphorylation of EGFR despite the inhibition of its downstream MEK/ERK1/2 and mTOR/S6K cascades (Figs. 3 and 7). Combination therapy consisting of BiP inhibitors and EGFR inhibitors resulted in more profound inhibitory effect on CAA and HCC cell lines growth than BiP inhibitors or EGFR inhibitors alone. We also tested the therapeutic efficacy of combined YUM70 and Osimertinib treatment in HCCC9810 cells without EGFR reactivation induced by YUM70. Consistently, the results also demonstrated significantly increased effectiveness of the combination therapy compared to the monotherapy. And these findings support the critical role of targeting ERK1/2 cascade in overcoming acquired resistance to EGFR TKI. But the mechanism of EGFR feedback activation after ERK1/2 blockade by BiP inhibitors should be fully clarified in our future study, which might provide a new target therapy strategy for CCA and HCC.
Taken together, our findings reveal novel mechanisms of the anti-tumor activity of YUM70 as one novel BiP inhibitor in CCA and HCC cells, which further supports the potential of YUM70 as an anticancer agent for CCA and HCC therapy. In the study, we firstly confirmed that YUM70 induced cell death mechanisms contained autophagy related apoptosis and GSDME dependent pyroptosis mediated by activated Caspase-3 in CCA and HCC cells. Until now, it is the first time that YUM70 induced pyroptosis in cancer cell lines has been reported. Secondly, the addition of lower concentrations of EGFR inhibitors abrogates EGFR reactivation and sensitizes CAA or HCC cells to BiP inhibitor treatment, resulting in a synergistic combination. Our work suggests that the addition of a low dose BiP inhibitor to standard TKI treatment may improve prognosis for patients with advanced CCA and HCC.
GSDME, gasdermin E; GRP78, 78-kDa glucose regulated protein; ER, endoplasmic reticulum; UPR, unfolded protein response; EGFR, epidermal growth factor receptor; TKI, inhibitors of tyrosine kinases; LC3, microtubule-associated protein 1 light chain 3; HCC, hepatocellular carcinoma; CCA, cholangiocarcinoma; EMT, epithelial-mesenchymal transition.
Financial disclosure statements:
This study was financially supported by the cultivation project for high-level talents of JiNan Central Hospital (No. YJRC2021007).
Author contributions
Lei Sun and Hao Zhou performed the experiments and YaNan Liu and MingYan Zhang helped analyze the data. Jian Zhang, XueLei Cao and Lei Sun designed the project. Lei Sun and XueLei Cao analyzed the data and wrote the manuscript. Jian Zhang and XueLei Cao revised the manuscript.
Acknowledgements
We thank Dr. MeiLi Sun (Central Hospital affiliated to ShanDong First Medical University) and Prof. HaoJie Jin (Shanghai Jiaotong University) for the suggestion of the project, and thank Prof. XiaoLu Zhang (ShanDong University) for providing HuCCT1 cell line.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
- Bray, F., et al., Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin, 2018. 68(6): p. 394-424.
- Marin, J.J.G., et al., Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers (Basel), 2020. 12(6).
- Rizvi, S., et al., Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol, 2018. 15(2): p. 95-111.
- Jin, H., et al., EGFR activation limits the response of liver cancer to lenvatinib. Nature, 2021. 595(7869): p. 730-734.
- Wu, B., Q.H. Sodji, and A.K. Oyelere, Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges. Cancers (Basel), 2022. 14(3).
- Vaquero, J., et al., The IGF2/IR/IGF1R Pathway in Tumor Cells and Myofibroblasts Mediates Resistance to EGFR Inhibition in Cholangiocarcinoma. Clin Cancer Res, 2018. 24(17): p. 4282-4296.
- Razumilava, N. and G.J. Gores, Cholangiocarcinoma. Lancet, 2014. 383(9935): p. 2168-79.
- Moeini, A., et al., Molecular Pathogenesis and Targeted Therapies for Intrahepatic Cholangiocarcinoma. Clin Cancer Res, 2016. 22(2): p. 291-300.
- Khaled, J., et al., Drug Resistance and Endoplasmic Reticulum Stress in Hepatocellular Carcinoma. Cells, 2022. 11(4).
- Cubillos-Ruiz, J.R., S.E. Bettigole, and L.H. Glimcher, Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell, 2017. 168(4): p. 692-706.
- Urra, H., et al., Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer, 2016. 2(5): p. 252-262.
- Hetz, C., K. Zhang, and R.J. Kaufman, Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol, 2020. 21(8): p. 421-438.
- Sano, R. and J.C. Reed, ER stress-induced cell death mechanisms. Biochim Biophys Acta, 2013. 1833(12): p. 3460-3470.
- Lee, A.S., GRP78 Induction in Cancer: Therapeutic and Prognostic Implications. Cancer Research, 2007. 67(8): p. 3496-3499.
- Lee, E., et al., GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res, 2006. 66(16): p. 7849-53.
- Wang, M. and R.J. Kaufman, The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer, 2014. 14(9): p. 581-97.
- Chen, X. and J.R. Cubillos-Ruiz, Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer, 2021. 21(2): p. 71-88.
- Zhang, X., et al., GRP78 blockade overcomes intrinsic resistance to UBA1 inhibitor TAK-243 in glioblastoma. Cell Death Discov, 2022. 8(1): p. 133.
- Kosakowska-Cholody, T., et al., HKH40A downregulates GRP78/BiP expression in cancer cells. Cell Death Dis, 2014. 5(5): p. e1240.
- Cerezo, M., et al., Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer Cell, 2016. 29(6): p. 805-819.
- Marciniak, S.J., J.E. Chambers, and D. Ron, Pharmacological targeting of endoplasmic reticulum stress in disease. Nat Rev Drug Discov, 2022. 21(2): p. 115-140.
- Samanta, S., et al., The Hydroxyquinoline Analogue YUM70 Inhibits GRP78 to Induce ER Stress-Mediated Apoptosis in Pancreatic Cancer. Cancer Res, 2021. 81(7): p. 1883-1895.
- Wang, Y., et al., Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature, 2017. 547(7661): p. 99-103.
- Shi, J., et al., Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature, 2015. 526(7575): p. 660-5.
- Yu, P., et al., Pyroptosis: mechanisms and diseases. Signal Transduction and Targeted Therapy, 2021. 6(1): p. 128.
- Kobayashi, T., et al., Bexarotene-induced cell death in ovarian cancer cells through Caspase-4-gasdermin E mediated pyroptosis. Sci Rep, 2022. 12(1): p. 11123.
- Rogers, C., et al., Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun, 2017. 8: p. 14128.
- Sarrió, D., et al., The multifaceted roles of gasdermins in cancer biology and oncologic therapies. Biochim Biophys Acta Rev Cancer, 2021. 1876(2): p. 188635.
- Kesavardhana, S., R.K.S. Malireddi, and T.D. Kanneganti, Caspases in Cell Death, Inflammation, and Pyroptosis. Annu Rev Immunol, 2020. 38: p. 567-595.
- Liu, X., et al., Channelling inflammation: gasdermins in physiology and disease. Nat Rev Drug Discov, 2021. 20(5): p. 384-405.
- Hu, L., et al., Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis, 2020. 11(4): p. 281.
- Liao, X.-X., et al., Gasdermin E: A Prospective Target for Therapy of Diseases. Frontiers in Pharmacology, 2022. 13.
- Erkes, D.A., et al., Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov, 2020. 10(2): p. 254-269.
- Zhang, Z., et al., Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature, 2020. 579(7799): p. 415-420.
- Zheng, Z.Y., et al., STAT3β disrupted mitochondrial electron transport chain enhances chemosensitivity by inducing pyroptosis in esophageal squamous cell carcinoma. Cancer Lett, 2021. 522: p. 171-183.
- Yoon, J.H., et al., Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J Hepatol, 2004. 41(5): p. 808-14.
- Chen, S., et al., Targeted therapy for hepatocellular carcinoma: Challenges and opportunities. Cancer Lett, 2019. 460: p. 1-9.
- Lemmon, M.A. and J. Schlessinger, Cell signaling by receptor tyrosine kinases. Cell, 2010. 141(7): p. 1117-34.
- O'Rourke, C.J., P. Munoz-Garrido, and J.B. Andersen, Molecular Targets in Cholangiocarcinoma. Hepatology, 2021. 73 Suppl 1: p. 62-74.
- Arteaga, C.L. and J.A. Engelman, ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell, 2014. 25(3): p. 282-303.
- Wu, Q., et al., EGFR Inhibition Potentiates FGFR Inhibitor Therapy and Overcomes Resistance in FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov, 2022. 12(5): p. 1378-1395.
- Misale, S., et al., Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci Transl Med, 2014. 6(224): p. 224ra26.
- Pellat, A., J. Vaquero, and L. Fouassier, Role of ErbB/HER family of receptor tyrosine kinases in cholangiocyte biology. Hepatology, 2018. 67(2): p. 762-773.
- Ye, Q.H., et al., GOLM1 Modulates EGFR/RTK Cell-Surface Recycling to Drive Hepatocellular Carcinoma Metastasis. Cancer Cell, 2016. 30(3): p. 444-458.
- Garcia-Lezana, T., J.L. Lopez-Canovas, and A. Villanueva, Signaling pathways in hepatocellular carcinoma. Adv Cancer Res, 2021. 149: p. 63-101.
- Sun, L., et al., Beclin-1-independent autophagy mediates programmed cancer cell death through interplays with endoplasmic reticulum and/or mitochondria in colbat chloride-induced hypoxia. Am J Cancer Res, 2015. 5(9): p. 2626-42.
- Herr, I. and K.M. Debatin, Cellular stress response and apoptosis in cancer therapy. Blood, 2001. 98(9): p. 2603-14.
- Qi, J., et al., Ciclopirox activates PERK-dependent endoplasmic reticulum stress to drive cell death in colorectal cancer. Cell Death Dis, 2020. 11(7): p. 582.
- Rozpedek, W., et al., The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr Mol Med, 2016. 16(6): p. 533-44.
- B'Chir, W., et al., The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res, 2013. 41(16): p. 7683-99.
- Sarvani, C., D. Sireesh, and K.M. Ramkumar, Unraveling the role of ER stress inhibitors in the context of metabolic diseases. Pharmacol Res, 2017. 119: p. 412-421.
- Orzalli, M.H., et al., Virus-mediated inactivation of anti-apoptotic Bcl-2 family members promotes Gasdermin-E-dependent pyroptosis in barrier epithelial cells. Immunity, 2021. 54(7): p. 1447-1462.e5.
- Liao, X.X., et al., Gasdermin E: A Prospective Target for Therapy of Diseases. Front Pharmacol, 2022. 13: p. 855828.
- Shen, X., et al., Caspase 3/GSDME-dependent pyroptosis contributes to chemotherapy drug-induced nephrotoxicity. Cell Death Dis, 2021. 12(2): p. 186.
- Zhang, Z., et al., Caspase-3-mediated GSDME induced Pyroptosis in breast cancer cells through the ROS/JNK signalling pathway. J Cell Mol Med, 2021. 25(17): p. 8159-8168.
- Urra, H. and C. Hetz, A novel ER stress-independent function of the UPR in angiogenesis. Mol Cell, 2014. 54(4): p. 542-4.
- Qiao, Q., et al., Endoplasmic reticulum stress pathway PERK-eIF2α confers radioresistance in oropharyngeal carcinoma by activating NF-κB. Cancer Sci, 2017. 108(7): p. 1421-1431.
- Bakir, B., et al., EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol, 2020. 30(10): p. 764-776.
- Lamouille, S., J. Xu, and R. Derynck, Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol, 2014. 15(3): p. 178-96.
- Zhou, P., et al., NMIIA promotes tumor growth and metastasis by activating the Wnt/β-catenin signaling pathway and EMT in pancreatic cancer. Oncogene, 2019. 38(27): p. 5500-5515.
- Poulikakos, P.I. and D.B. Solit, Resistance to MEK inhibitors: should we co-target upstream? Sci Signal, 2011. 4(166): p. pe16.
- Lito, P., et al., Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell, 2012. 22(5): p. 668-82.
- Riganti, C., et al., The role of C/EBP-β LIP in multidrug resistance. J Natl Cancer Inst, 2015. 107(5).
- Salaroglio, I.C., et al., PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol Cancer, 2017. 16(1): p. 91.
- Becker, J.H., et al., CXCR7 Reactivates ERK Signaling to Promote Resistance to EGFR Kinase Inhibitors in NSCLC. Cancer Res, 2019. 79(17): p. 4439-4452.
- Li, Y., et al., ERK inhibition effectively overcomes acquired resistance of epidermal growth factor receptor-mutant non-small cell lung cancer cells to osimertinib. Cancer, 2020. 126(6): p. 1339-1350.
- To, C., et al., An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nat Cancer, 2022. 3(4): p. 402-417.
- Frankson, R., et al., Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer Res, 2017. 77(21): p. 5701-5705.
- Bruner, J.K., et al., Adaptation to TKI Treatment Reactivates ERK Signaling in Tyrosine Kinase-Driven Leukemias and Other Malignancies. Cancer Res, 2017. 77(20): p. 5554-5563.
- Maik-Rachline, G., A. Hacohen-Lev-Ran, and R. Seger, Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int J Mol Sci, 2019. 20(5).
- Han, T., et al., PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J Hepatol, 2015. 63(3): p. 651-60.
- Park, K.S., et al., The HSP90 inhibitor, NVP-AUY922, attenuates intrinsic PI3K inhibitor resistance in KRAS-mutant non-small cell lung cancer. Cancer Lett, 2017. 406: p. 47-53.
- Xu, H., et al., Alpelisib combination treatment as novel targeted therapy against hepatocellular carcinoma. Cell Death Dis, 2021. 12(10): p. 920.
- Hsieh, C.L., et al., A Novel Salicylanilide Derivative Induces Autophagy Cell Death in Castration-Resistant Prostate Cancer via ER Stress-Activated PERK Signaling Pathway. Mol Cancer Ther, 2020. 19(1): p. 101-111.
- Lu, W. and Y. Kang, Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell, 2019. 49(3): p. 361-374.
- Shibue, T. and R.A. Weinberg, EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol, 2017. 14(10): p. 611-629.
- Bhattacharyya, S., et al., Decline in arylsulfatase B expression increases EGFR expression by inhibiting the protein-tyrosine phosphatase SHP2 and activating JNK in prostate cells. J Biol Chem, 2018. 293(28): p. 11076-11087.
- Nastiuk, K.L. and J.J. Krolewski, Opportunities and challenges in combination gene cancer therapy. Adv Drug Deliv Rev, 2016. 98: p. 35-40.
- O'Reilly, K.E., et al., mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res, 2006. 66(3): p. 1500-8.
- Colli, L.M., et al., Landscape of Combination Immunotherapy and Targeted Therapy to Improve Cancer Management. Cancer Res, 2017. 77(13): p. 3666-3671.
No competing interests reported.
- floatimage9.png
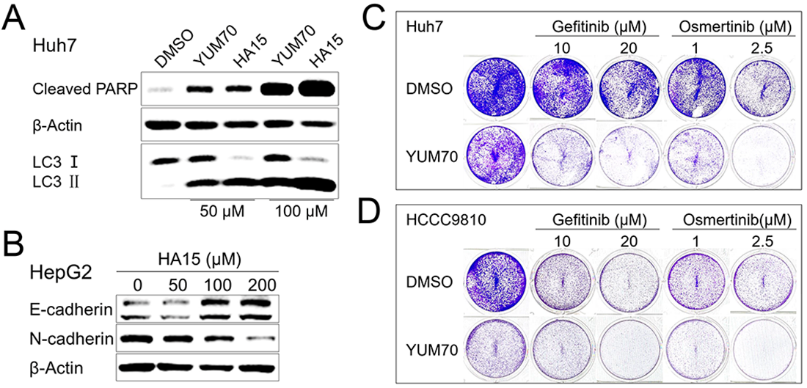
Supplement 1 (A) Immunoblot analysis of Cleaved PARP, LC3 and β-Actin in Huh7 upon treatment with 50 μM and 100 μM YUM70 or HA15 for 24 hours. (B) E-cadherin, N-cadherin and β-catenin proteins were detected by western blotting in HepG2 cells treated with increasing concentration of YUM70 (DMSO, 50, 100, 200 µM) for 24 hours. (C and D) Huh7 or HCCC9810 cell lines in 12-well plates were exposed to DMSO, 10 or 20 μM Gefitinib, 1 or 2.5 μM Osimertinib, 50 μM YUM70 or the indicated combinations. After 48 h, the cells were fixed and stained. Representative staining images are shown.
{kind=link}