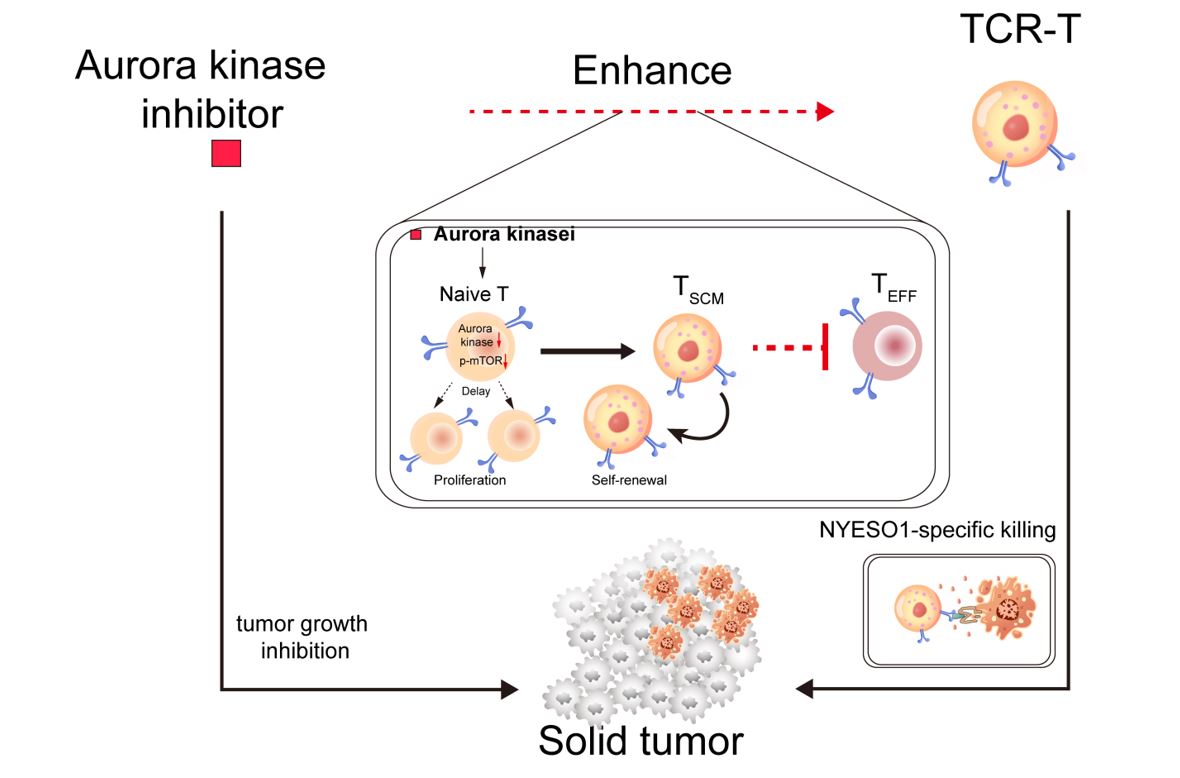
Aurora kinase inhibitors (AURKi) -induced TCR-T cells lead higher recall response and longer lasting anti-tumor immune effect
Since combining small molecular inhibitors with T cell therapy is proven to overcome the limitation faced by T cell therapy alone [31]. To determine whether AURKi own potency to enhance TCR-T cells, we developed an in-vitro combination approach by adding AURKi (Alisertib and Barasertib-HQPA) in TCR-T cell preparation process.
The potency to produce more antigen-specific effector T cells is essential to determine in vivo immune response of T cell therapy [32]. Therefore, we first examined the recall response and antitumor effect of AURKi-treated TCR-T cells in an ACT tumor model. TCR-T cells were produced from isolated naïve T cells. Isolated human T cells were incubated with 10 µM of Alisertib - an aurora A inhibitor (AURKAi), and 10 nM of Barasertib-HQPA - an aurora B inhibitor (AURKBi), at the time the cells were activated, followed by specific lentivirus infection.
Then the NY-ESO-1-specific TCR-T cells were amplified in the presence of IL-2 for another 4 days. The viability of T cells was not significantly affected at the concentration of both drugs even for 8 days of incubation (Fig. S1A, B). We also determined the specific functionality of the harvested TCR-T cells. Upon stimulation of NY-ESO-1 peptides and A375 cells (NYESO1+HLAA2+), expression of effector function markers (IFN-γ, GZMB) and cytotoxicity in AURKi (AURKAi, AURKBi)-treated TCR-T cells were similar to that in control TCR-T cells (Fig. S1C-E), indicating that drugs involvement would not significantly affect specific TCR transduction and expression.
After being re-stimulated with NY-ESO-1 peptides for another 2 days, both AURKAi-TCR-T cells and AURKBi-TCR-T cells produced more CD62L-CD44 + effector T cells than control TCR-T cells (Fig. 1A,B), indicating a stronger antitumor potency. We next tested the immune effects of AURKi-TCR-T cells, in a subcutaneous tumor model that carry specific human antigens, NY-ESO-1 (Fig. 1B).
Upon tumor formation, mice were either treated with or without TCR-T cells. And the TCR-T cells were pretreated with or without AURKi. AURKi, especially AURKBi significantly enhanced antitumor response of TCR-T cells and revealed a prolonged overall survival (Fig. 1C-E). As the results showed, all mice survived in the AURKBi-TCR-T group, while only 60% and 0% survived in other TCR-T groups and no treatment group, respectively (Fig. 1E). Following FACS analysis of tumor infiltrated lymphocytes (TILs) on day 10 after treatment, we found that total CD8+T cells in AURKi-TCR-T groups, both AURKAi- and AURKBi-TCR-T groups, significantly exceeded those in conventional TCR-T group. And AURKBi pretreatment produced even more CD8+ TILs than AURKAi pretreatment (Fig. 1F). A significant increased percentage of CD8+T cells in AURKAi-TCR-T and AURKBi-TCR-T expressed activation markers (IL2Rβ and CXCR5) than those in conventional controlled TCR-T groups were observed (Fig. 1G). We also found an increase in granzyme B and IFN-γ expressed TILs, while an decrease in CD8+ T cells expressing inhibitory receptors (LAG3) (Fig. 1H, I), indicating that transferred AURKi-TCR-T cells expanded to functional effector T cells with less exhausted state.
The increased functional effector T cells may benefit from stronger immume memory, as this was confirmed by the observation that more CCR7+, TCF1+ and CD62L+ TILs in AURKi-TCR-T groups compared with those in controlled TCR-T group (Fig. 1J, K). Furthermore, AURKBi-TCR-T group showed higher potency to produce T cells with stem memory phonotypes than AURKAi-TCR-T group did, therefore, persistently generated more CD44+ effector T cells even on day 40 after treatment (Fig. 1L). Together, these data show that AURKBi potentially enhances antitumor effects of TCR-T cells, by in-vivo generating more less-exhausted, activated effector T cells, which may drive by larger number of TCR-T cells maintained their stem memory phenotypes.
Dose- and time-dependent AURKB inhibition generates less-exhausted TCR-T cells
Given the in-vivo generation of more stem-like and less-exhausted T cells due to transferred of AURKBi-treated TCR-T cells, we further assessed whether in-vitro AURKB inhibition is directly implicated in the increase of stem memory related markers. To this end, differently concentration of AURKBi was integrated into TCR-T cell preparation, and following expression of CD62L and CCR7 in T cells were analyzed. We found that in a specific range (0–20 nM), the percentage of CD62L- and CCR7-positive CD8 + T cells were elevated (Fig. 2A), suggesting that indeed AURKB inhibition lead to maintenance of naïve- and memory-like properties, and this partially depended on the dose of AURKBi. Next, we used 10 nM and 20 nM of AURKBi, which showed the most significant inducing effect, to address TCR-T cells followed by further investigation of states and phenotypes of the harvested TCR-T cells (Fig. 2B). After IL-2 induced extension, TCR-T cells without AURKBi treatment (Ctrl) or with AURKBi at both concentrations formed proliferation aggregates. Despite that 20 nM of AURKBi leaded to slightly smaller aggregates (Fig. 2C), T cell proliferative ability was not significantly affect in these treatments, as determined by the CFSE labelling (Fig. 2D). Furthermore, we characterized AURKBi-induced TCR-T cells for their memory formation. The proportion of TCM (CD62L+CD44+) cells from AURKBi-induced TCR-T cells was similar to that from control TCR-T cells, while the presence of AURKBi resulted in a significant increase in a naïve like phenotype (CD62L+CD44−) (Fig. 2E). However, unlike naïve cells, these AURKBi-induced TCR-T cells showed higher expression of activation markers (CXCR5 and IL-2Rβ) (Fig. 2F, G). Besides, these naïve-like T cells expressed significantly lower levels of exhaustion (LAG3) (Fig. 2H) and effector function-associated mRNAs (IFNg, PRF1) (Fig. 2I, J). Besides having higher expression of memory (CCR7) marker, AURKBi-induced TCR-T cells also showed upregulated expression of TCF7 and Bcl2 (Fig. 2K, L), which are associated with increased stemness and prolonged survival [33–35]. In addition, these CD62L+CD44− T cells in the AURKBi-treated groups showed significant higher proliferative ability (Fig. 2M). And AURKBi-treated T cells also showed reduced apoptosis, as confirmed by lower fluorescence intensity of annexin V (Fig. 2N).Together, AURKBi treatment induced a subgroup of T cells sharing both naïve and memory characteristics. Earlier, a population of less differentiated T cells with stem memory properties, possessed intermediate state between naïve T cells and TCM cells, has been described [36]. Hence, these data together suggested that AURKB inhibition promoted production of a subset of T cells that are more like stem memory T (TSCM) cells.
Above observations prompted us to test whether this inducing effect becomes stronger with extended co-incubation time, and further optimize the treatment. We compared percentage of CD62L+CD44− T cells between two preparation programs (Fig. 3A). The results showed that late removal of AURKBi (program 2) increased CD62L+ T cell population than early removal (program 1) (Fig. 3B, C), indicating the time-dependent effect of exhibit AURKBi induction. However, the total number of cells harvested from program 2 was less than that from program 1 ((Fig. 3D). Despite this, TCR-T cells generated by program 2 exhibited more potent tumor suppression effects and extended survival (Fig. 3E, F). All these data suggested that AURKB inhibition assisted T cells maintained their stem-like phenotypes in a dose- and time-dependent manner, therefore, generated less-exhausted TCR-T cells. Although longer co-incubation of AURKBi slightly attenuated cell extension, it enhanced persistence of TCR-T cells in vivo.
AURKB inhibition mediated enhancement in FAO that maintains stem memory properties
To further characterize TSCM properties of AURKBi-induced TCR-T cells, we added AURKBi during the whole procedure of TCR-T cells preparation and expansion (Fig. 4A), and observed the harvested TCR-T cells by TMRE (tetramethylrhodamine, ethyl ester), which is used for quantifying changes in mitochondrial membrane potential. We observed lower fluorescence intensity of TMRE in TCR-T cells treated by AURKBi, which acts as a factor of lower mitochondrial potential, a further indication of stem-like characteristics [37]. And 20 nM of AURKBi acquired more significant decrease than 10 nM of AURKBi did (Fig. 4B), again demonstrated this dose-dependent inducing effect by AURKBi treatment in vitro.
Enhanced fatty acid oxidation (FAO) is a strong indicator of memory and long-term cell survival of T cell, and is also a critical factor for stem cell –like memory T cells [38]. Therefore, we next determined the status of FAO in the AURKBi-treated TCR-T cells. AURKBi-mediated TCR-T cells displayed elevated production of β-hyroxybutyrate (BHBA) (Fig. 4C), the final production of fatty acid β-oxidation [39]. In line with this, we found increased and increased expression of CPT1A (Fig. 4D), a rate limiting enzyme of FAO [38], thus showing enhancement of FAO by AURKBi-treated TCR-T cells. Furthermore, we found SIRT3, the downstream of PGC1α [40], was also increased in TCR-T cells after AURKBi inhibition (Fig. 4E, F). In contrast, a suppression effect on Glut1 and HK2 was observed in AURKBi-mediated TCR-T cells (Fig. S2). As a consequence, these TCR-T cells weakened their glycolytic metabolism, resulting in reduced levels of lactate in the culture supernatants (Fig. 4G). These data showed that AURKB inhibition promotes FAO and decreases glycolysis.
To further confirm that enhanced FAO is correlated to TSCM inducing, we tested whether the increase of TSCM properties related to AURKB inhibition is attenuated in the context of the combination treatment with etomoxir (Fig. 4H), an inhibitor of CPT1A. The expression of CD62L and CCR7 on T cells, the indicators of naïve and memory phenotypes, were markedly decreased in TCR-T cells with combined treatment compared with those treated with AURKBi alone (Fig. 4I, J). Besides, effector markers (IFN-γ and GZMB) were lower while exhausted marker (LAG3) was higher in terms of combination treatment with etomoxir (Fig. S3). Altogether, these results demonstrated that AURKBi induced TSCM cells by reprogramming glycolytic-based into FAO-based metabolic programme.
AURKBi induces TSCM cells by decreasing cell proliferation that depends on controlling mTOR substrates
Although short-term incubation of AURKBi did not significantly affect cell proliferation (Fig. 2B), it does maintained TSCM attributes. Therefore, we next determined how AURKBi affect cell differentiation in this short-term manner (Fig. 5A). We found that the majority of the cells differentiated to effector T (TEFF) and central memory T (TCM) cells, while AURKB inhibition enriched TSCM cells even in the early generations (generations 0 and 1). Indeed, the proportion of TSCM increased in the new generations (generations 2 and 3), especially when higher dose of AURKBi (20 nM) were involved (Fig. 5B, C). These results confirmed the dose-dependent effect of AURKBi, and prompted us to further determine whether AURKBi affect cell proliferation and differentiation cycle in time-dependent manner. We observed long- term AURKB inhibition significantly decreased cell proliferation (Fig. 5D, E).
Given the fact that retroviruses (including lentivirus) exclusively infect dividing cells [41], we tested the expression of Vβ8, which indicated transduction efficiencies [42], between control TCR-T cell and the TCR-T cell mediated by AURKBi. Surprisingly, only marginal differences in transduction efficiencies were observed. The transduction efficiencies were relatively high for all groups, about 72%, 70% and 65% separately (Fig. 5F). Despite that restraining T-cell expansion and slightly reducing the transduction efficiency, long-term and high-dose of AURKB inhibitions generated about 4-fold higher expression of CD62L and CCR7, which indicating TSCM properties, than those without AURKBi (Fig. 5G-K). AURKBi caused an increased accumulation of cells in the earlier generations of cell division, indicating a delay in the cell cycle progression. But AURKB inhibition does not significantly affect T cell activation, as was confirmed by similar expression levels of p-PI3K and p-Akt (Fig. 5L, M), which involved in mediating T cell activation [13]. mTOR signal is critical to promote cell cycle progression and cell growth [43]. Earlier, it has been suggested that aurora B is required to fully activated 4E-BP1, a downstream substrate of mTOR, and enhanced phosphorylation of mTOR [30, 44]. Accordingly, we checked the effect of AURKBi on phosphorylation of mTOR and 4E-BP1. As anticipated, in the presence of AURKBi, phosphorylation of mTOR and 4E-BP1 was suppressed (Fig. 5N, O, and Fig. S4), indicating AURKB inhibition directly or indirectly control the activity of mTOR signal. Hence, AURKBi delay cell cycle progression and inhibited their further differentiation without affecting activation of T cells.
Besides, previous studies have demonstrated that MEK and GSK-3β inhibitors involving in the activation of naïve T cells can generate TSCM cells [13, 45], suggesting that it is the unactivated T cells that be induced into TSCM cells during their priming process. In this study, we also determine the state of T cells that can be more likely to generate TSCM cells. In line with previous studies, naïve T cells were robustly reprogrammed into TSCM cells in their priming process while treated with AURKBi. However, the control TCR-T cells, although still produced TSCM cells while being treated with AURKBi during second stimulation, generated much less number of TSCM cells than those treated with AURKBi during the priming procedure (Fig. S5). This observation may attribute to the fact that the proportion of naive T cells decreased in preactivated T cells.
AURKBi-induced TCR-T treatment in BALB/C-nude mice with metastatic tumor from human melanoma
Since we demonstrated that in a long-term and high-dose manner, AURKB inhibition could reprogram TCR-T cells with more TSCM properties, which may be more potent than conventional TCR-T cells in treatment of solid tumor. Finally, we established a new method for preparing improved TCR-T cells by preparing TCR-T cells for a total of 7 days commitment with incubation of 20 nM of AURKBi. To demonstrate the superiority of the TCR-T cells produced by the method, we first initiated the in vivo studies with metastatic melanoma model, which is more challenged to be treated than subcutaneous model.
The tumor model was established by injecting A375 cells from tail veils (Fig. 6A). After a week, tumor nodes were observed in lung (Fig. 6B), and mice were randomly divided into three groups: no treatment control, conventional TCR-T, and AURKBi-TCR-T. We detected a stronger suppression in tumor growth in mice that received AURKBi-TCR-T cells as compared to no treatment control and conventional TCR-T groups (Fig. 6C). Mice that received AURKBi-TCR-T cells also had a significantly prolonged survival (Fig. 6D). Given that fresh active T cells with effective anticancer activity is largely provided by peripheral tissues [46], we compared the number and state of T cells from blood and spleen between conventional and AURKBi-TCR-T groups, at day 17 and 29 after tumor cell injection. At 17 day, we found that the two cell populations (conventional or AURKBi- TCR-T) had equal levels of T cells in blood and spleen (Fig. 6E, F), and these T cells expressed equal level of exhausted markers (PD1 and LAG3), effector function marker (IFN-γ) and activation markers (CXCR5 AND IL-2Rβ) (Fig. S7). But AURKBi- TCR-T group showed higher expression of stem memory makrers-CCR7 and CD62L (Fig. 6G and Fig. S7), suggesting that AURKBi- induced TSCM cells kept in a relative rest state without meeting large number of tumor antigens in peripheral tissues. However, the population of total T cells and CD62L+ T cells in blood and spleen decreased at day 29 (Fig. S8), partially due to that they were recruited into the tumor. This was confirmed by the observation that AURKBi-TCR-T cell transfer presented higher frequencies of CD8 + T cells in the tumor, at day 29, compared to conventional TCR-T cell transfer (Fig. 6H). Furthermore, intratumoral AURKBi-treated CD8+ T cells showed higher expression of CD62L, CD44, CXCR5 and IL2-Rβ expression (Fig. 6I, J and Fig. S9), indicating maintenance of stem-like phenotype and production of more activated effector T cells at the same time in AURKBi-treated CD8+T cell compared to non- AURKBi-treated cells. In addition, AURKBi-treated CD8+T cells exhibited equal expression of PD-1, with a significantly lower expression of LAG3 compared to non- AURKBi-treated cells, showing effectively activated and less-exhausted state in AURKBi-treated CD8 + T cells (Fig. 6K, L). These result demonstrated that AURKBi-induced TCR-T cells maintained TSCM population in vivo that played as reservoir for effective tumor infiltrated T cells, which showed potential to effectively suppress metastatic tumor.
{kind=link}