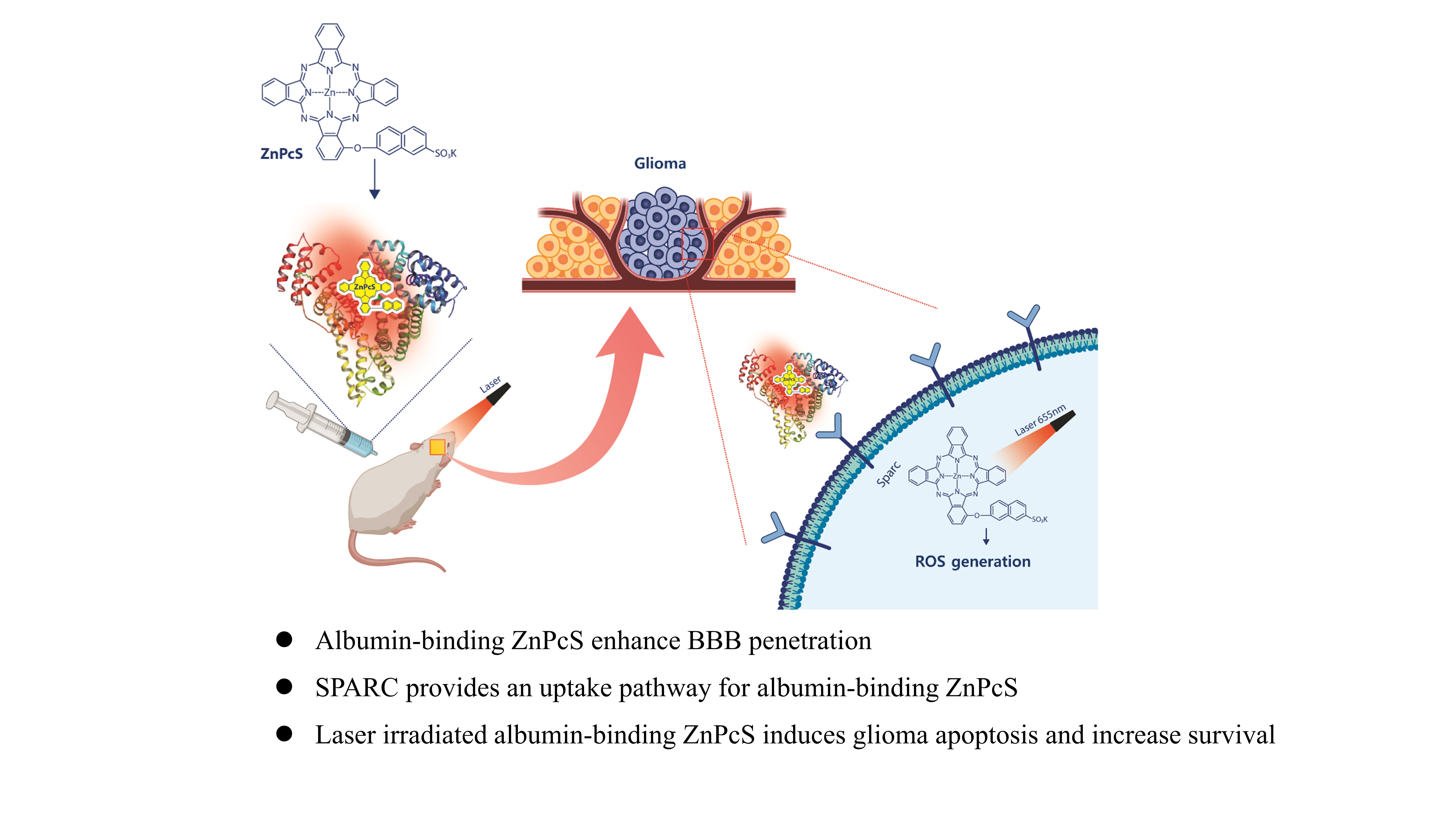
Chemical materials and instruments
Potassium 6-hydroxy-2-naphthalenesulfonate, phthalonitrile, 3-nitrophthalonitrile, potassium carbonate, zinc acetate, dimethyl sulfoxide (DMSO), n-pentanol, 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU), and 2,7-dichlorofluorescin diacetate (DCFH-DA) were purchased from Sigma-Aldrich, Korea. High-resolution mass spectra were obtained using Exactive Plus Orbitrap (Thermo Fisher Scientific). Proton nuclear magnetic resonance spectra were recorded on a Bruker 300 MHz. Fluorescence spectra were collected using an Edinburgh FL900/FS900 spectrofluorometer. Electronic absorption spectra were recorded using a SHIMADZU UV-2450 spectrophotometer.
Molecular Modeling Studies
Protein and ligand preparation. The X-ray crystal structures of human serum albumin (HSA) bound to heme molecules (PDB codes:1N5U3 and 1O9X4) were prepared using the Protein Preparation Wizard in Maestro, version 9.2 (Schrödinger, LLC, NY, USA) [24, 25]. Bond orders were assigned, hydrogen atoms were added, and protonation states of the residues at pH 7.4 were generated by Epik, version 2.6. All hydrogen atoms were energy-minimized with the optimized potential for liquid simulation (OPLS) 2005 force field until the average root-mean-square deviation in the hydrogen atoms reached 0.30 Å. Metal coordination in the ZnPcS ligand was prepared using the protein preparation wizard. The resulting structures were energy-minimized using the implicit solvent model and OPLS2005 force field.
Blind docking and binding mode selection. The ligands were docked into several binding pockets of HSA using Glide, version 6.1, in Maestro, according to the following protocol: i) The grid box was generated using the centroid of the co-crystallized ligand. To ensure that all the binding cavities in the HSA were covered, we set the grid box size to be sufficiently large. ii) Glide standard precision docking was used for the docking stage, and up to 500 conformations of each ligand were identified. iii) The docked conformations were clustered based on their root-mean-square deviations, and the final modes were chosen based on their scores and population.
The computational tasks were performed on a Linux CentOS 5.8 workstation with an Intel Xeon octa-core 2.5 GHz processor, and molecular graphic figures were generated using PyMOL software (http://www.pymol.org).
Cell Culture
The human glioma U87 cell line was purchased from the American Type Culture Collection (ATCC, USA) and cultured in Eagle’s Minimum Essential Medium with 1.5 g L− 1 sodium bicarbonate, nonessential amino acids, L-glutamine, sodium pyruvate, and 1% Penicillin/Streptomycin (Corning, NY, USA) with 10% heat-inactivated fetal bovine serum (FBS, GWVITEK, Seoul, Korea). The human umbilical vein endothelial cell (HUVEC) cell line was purchased from ATCC and cultured in EGM2 medium with supplements. Confluent cells were passaged every 4–5 days using 0.05% trypsin/ethylenediaminetetraacetic acid. All the cells were maintained in an incubator at 37°C with 5% CO2 in a humidified atmosphere.
Immunocytochemistry
To demonstrate the accumulation of ZnPcS in glioma cells, immunocytochemistry was performed using confocal microscopy (LSM710; Carl Zeiss Microimaging GmbH, Jena, Germany). Briefly, the cells were cultured in a 35 mm confocal microscope dish for 48 h and then treated with ZnPcS at the indicated concentrations. After the indicated incubation times, the cells were first washed in PBS and then fixed with 4% paraformaldehyde for 20 min. After a series of washes with PBS, the 4’,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, Waltham, MA, USA) was treated to make the nuclear contrast. To identify the protein expression of SPARC, the SPARC antibody was applied to fixed cells. After washing twice with PBS, the samples were blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature and washed again with PBS. The cells were incubated with the primary antibody overnight. The dishes were washed thrice with PBS/0.05% Triton X-100 and incubated with the secondary antibody for 2 h at room temperature in the dark. After washing with PBS/Triton X-100, the cells were counterstained with DAPI. All the fluorescence images were captured using a Carl Zeiss confocal laser scanning microscope with a 20× objective.
Photo-induced Cytotoxicity Against U87-glioma
The effect of ZnPcS on U87-glioma viability was measured using the MTT assay. Briefly, 1×104 cells were seeded in 96-well tissue culture plates. After 24-h incubation, the cells were treated with different concentrations of ZnPcS (0–20 µM in MEM medium) for 24 h. The cells were recharged with fresh medium, exposed to laser irradiation (655 nm, 200 mW, 1 min), and further incubated for 24 h. Cell viability was evaluated using the MTT assay (Theranostics 2019). MTT reagent was added to each well, and the cells were incubated for 4 h. The DMSO was added to dissolve the formazan crystals, and the absorbance of the purple formazan was measured at 570 nm using a microplate reader.
Intracellular Ros Detection
ROS generation within the cells was identified using DCFH-DA as a ROS indicator by means of fluorescence. The cell-permeable non-fluorescent DCFH-DA can freely cross the cell membrane to enter the cell and can be oxidized to form a green fluorescent DCF. To identify ROS generation in culturing U87-glioma, the cells were cultured in a confocal culture dish. After one hour of ZnPcS treatment of U87, the cells were subjected to laser irradiation for 1 min. The cells were washed thrice, and the culture medium of DCFH-DA (10 µM) was further incubated with the cells for 30 min (biomacromolecules).
Penetration of ZnPcS using in vitro BBB model
To determine whether ZnPcS can penetrate the BBB, an in vitro BBB model using a Transwell was used. The human endothelial cell line HUVEC was seeded in a Transwell upper chamber (Corning, USA) for monolayer cell culture. The cells were cultured for two days to become fully confluent. The U87-glioma was seeded in the lower chamber. The upper chamber was treated with ZnPcS, and the U87-glioma cells in the lower chamber were collected and washed with PBS to obtain fluorescence images.
In vivo of intracranial glioma xenograft model
Five-week-old female Balb/c nude mice were purchased from Nara Biotech (Seoul, Korea), and one week later, we started the experiment with six-week-old mice under specific pathogen-free conditions. To briefly explain the entire process, orthotopic glioma tumor mouse models were developed by transplanting Luc-U87 (stable transfection of firefly luciferase) into the left cerebral hemisphere using a stereotaxic apparatus (Harvard, MA, USA). The skull was leveled between the bregma and lambda. A small hole was drilled at the desired location and 1×106 Luc-U87M cells in 3 µL of PBS were injected into the frontal lobe based on the following coordinates (relative to Bregma) using a Hamilton syringe (10 µL Gastight Syringe Model, 1701 RN, Small Removable Needle, 26s gauge, 2 in, point style 2, Reno, Hamilton Company, NV, USA, Nevada, USA): 2.7 mm ventral from the dorsal surface of the skull, 0.5 mm caudal, and 1.1 mm lateral. The rate at which cells enter the brain was set to 0.5 µL/min, and the cells were injected. The growth of orthotopic glioma was monitored using bioluminescence imaging performed in the IVIS imaging system by intraperitoneal injection of luciferin substrate (150 mg/kg) [17].
To follow the ZnPcS distribution in vivo, fluorescence was measured in the major tissues after the IP administration of ZnPcS. After treatment with ZnPcS, the mice were euthanized to collect major tissues, including the brain, for fluorescence imaging at 1 h. The mice had continuous access to sterilized food pellets and distilled water and were housed under a 12 h light-dark cycle (7:00 am–7:00 pm) with a temperature of 23°C ± 1–2, and humidity of 50%±5. All the animal experiments were approved by the Institutional Animal Care and Use Committee of Soon Chun Hyang University and performed in accordance with the guidelines of the Experimental Animal Center of Soonchunhyang Institute of Medi-Bio Science.
In vivo antiglioma therapy
Mice with intracranial gliomas were randomly divided into five groups. The growth of orthotopic gliomas was monitored using luciferin for in vivo imaging. Treatment with ZnPcS was conducted seven days after Luc-U87 implantation, and the brain was irradiated by a 600 J laser. After irradiation, the growth of the orthotopic glioma was measured by the intensity of fluorescence, and the glioma size was monitored every week using IVIS. The survival rate and median survival time were calculated, and statistical differences were assessed using the Kaplan–Meier method.
Image Analysis
Anesthetized mice were placed in a light-tight chamber of Living Image version 4.5 (PerkinElmer, USA). For in vivo imaging, the mice were anesthetized with isoflurane (Hana Pharm, South Korea). Luciferase expression at the tumor site was visualized with the injection of luciferin (XenolightTM D-Luciferin Potassium Salt, 1 g, PerkinElmer, Waltham, MA, USA) and imaged by IVIS, and the luminescence intensity represented the tumor progression in the brain.
The mice were euthanized under isoflurane anesthesia prior to tissue collection via thoracotomy for biodistribution. Pseudo-color images indicating photon counts were overlaid on the images of the mice using Living Image software v. 2.25 (Caliper). A region of interest was manually selected based on the signal intensity. The area of the region of interest was kept constant, and the intensity was recorded as the maximum radiance within each region of interest [26].
Sparc Knock-out
The CRISPR/CAS9 SPARC Knockout Kit (ORIGENE, KN409964) was used for SPARC knockout. The SPARC gRNA CRISPR vector (KN409964G1, KN409964G2) and non-homology-mediated-based linear donor DNA (KN409964D, EF1a-Luciferase-P2A-Puro) were transfected by nucleofection. Transfections were performed using the Amaxa NucleofectorTM system (SE Cell Line 4D-NucleofectorTM X Kit L, LONZA), according to the supplier’s recommendations. For each cell, a control transfection with the pmax-GFP (provided with the Amaxa Nucleofector kit) plasmid alone, and co-transfection with the pmax-GFP plasmid along with the SPARC gRNA CRISPR plasmid and donor DNA, was performed. For each transfection, cells were resuspended in 100 µL nucleofector solution, 1 µg of each plasmid and donor DNA was added, and electroporation settings (program: DS126, CA137) were applied and then seeded in six-well plates with 2 ml growth media. Transfected cells were grown in growth media for 11–48 h until the majority of the cells showed visible GFP expression. After sufficient growth, SPARC-knockout cells were selected using puromycin (Gibco).
Western Blotting
Western blotting
Whole cell lysates were extracted using NP40 (Elpis Biotech, Daejeon, Korea) with a 100× protease inhibitor cocktail (Cell Signaling Technology, Danvers, MA, USA). The total protein concentration in each sample was measured using the Bradford assay. The protein samples were separated by electrophoresis using 12% SDS-polyacrylamide gels and transferred to 0.2-µM polyvinylidene fluoride blotting membranes (Amersham, Little Chalfont, UK). The membranes were blocked for 1 h in TBS-T (50 mM Tris, 0.15 M sodium chloride, 0.05% Tween 20) containing 5% blocker (BioShop, Burlington, Canada) and hybridized to primary antibody overnight at 4°C. The primary antibodies used were SPARC (Santa Cruz Biotechnology, Dallas, TX, USA) and β-actin. The membranes were washed thrice for 15 min with TBS-T at room temperature and incubated for < 1.5 h at room temperature with secondary antibodies. Chemiluminescence detection was performed using the Pico EPD Western Blot Detection Kit (Elpis Biotech) [27].
{kind=link}