Synthesis of new anticancer candidates with protein kinases inhibitory potency is a major goal of pharmaceutical science and synthetic research. This current work represents the synthesis of a series of substituted thiazolidinones incorporating a benzoate moiety, starting from 4-formylphenyl benzoate 1a and 4-formyl-2-methoxyphenyl benzoate 1b. Most prepared thiazolidinones 5a-j, 7a-h and 9a-j, were evaluated in vitro for their potential anticancer activity against three cell lines (HepG2, MCF-7 and HeLa). The most active cytotoxic compounds 3a, 3b, 5a, 5c and 5h were then further tested against the normal cell line WI38. All of these were shown be more effective toward anticancer cell lines. Thiazolidinones 5c and 5h were further evaluated to be kinase inhibitors against EGFR showing effective inhibitory impact. Furthermore, 5c and 5h were tested for their effects on cell cycle and apoptosis induction capability in HepG2 cell lines by DNA-flow cytometry analysis and annexin V-FITC apoptosis assay, respectively. The results showed that they have effect of disrupting the cell cycle and causing cell mortality by apoptosis in the treated cells. Moreover, molecular docking studies by the Moe 2015 program showed better binding patterns for 5c and 5hwith the active site of the EGFR protein kinase [PDB code 1M17]. Finally, toxicity risk and physicochemical characterization was performed for most of the compounds, revealing excellent properties as possible drugs, especially compounds 5c and 5h.
Research Article
Synthesis and molecular docking study of new thiazolidinones incorporating a benzoate moiety as anti-HepG2 cancer agents, EGFR inhibitors and apoptosis inducers
https://doi.org/10.21203/rs.3.rs-2444022/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Thiazolidinones
EGFR protein kinase
cell cycle DNA-flow cytometric
Annexin V-FITC
Cancer is a major problem affecting people`s lives worldwide, threatening both developed and developing countries and causing high ratio in death incidence. [1] Its incidence is reported to reach about 29.5 million cases each year in 2040. [2, 3] Liver cancer ranks sixth in the world among all malignancies. It’s considered the third most deadly cancer type among all. [4] It is estimated that by 2025, more than 1 million people will be diagnosed with liver cancer annually. [5] Hepatocellular carcinoma (HCC) is the most occurring type of liver cancer and causing about 90% of cases. Unfortunately, chemotherapeutic agents used to treat cancer are not selectivity other than their toxicity and resistance. [6] Therefore, extensive efforts has been devoted to developing new drugs that target only cancer cells. [7] Current drug discovery research targets protein kinases [8], which play critical role in cell proliferation and mobility so any dysfunctions occurred because of kinases mutations lead to cellular abnormalities and successively developing of cancer initiation, progression or even metastasis. [9] As such, protein kinases are considered as drug targets for chemotherapeutic agents, one of which is EGFR. [10–12]
Epidermal growth factor receptor (EGFR) is a trans-membrane glycoprotein that belong to the category of receptor tyrosine kinases and plays a key role in cell signaling pathways including cell proliferation, apoptosis, angiogenesis and metastatic invasion. [13] High-levels of his EGFR due to its overexpression are found in many types of human cancers, such as hepatocellular carcinoma and brain cancer, so EGFR is a remarkable target in the treatment of different types of cancer. [14–18] Late-stage of hepatocellular carcinoma shows, increased EGFR abundance that leads to increased proliferation and tumor differentiation. [19, 20] Recent studies on EGFR inhibitors in human hepatocellular carcinoma cell lines have enhanced our understanding of EGFR signaling mechanism in hepatocellular carcinoma and thus will improve the application of these inhibitors in liver cancer therapy. [21–25] The EGFR pathway is inactivated by tyrosine kinase inhibitors (TKIs) like Gefitinib and Erlotinib (Fig. 1). They are antagonists of binding to the EGFR adenosine triphosphate pocket, in turn inactivating EGFR auto-phosphorylation and downstream signaling. [26–29] Reportedly, resistance to cancer therapy arising from anticancer drugs, radiotherapy and even hormonal therapy was caused by over-expression of EGFR in variety of human cancers. [30, 31] Therefor, in his case with advanced EGFR mutations, anti-EGFR drugs are the first line of treatment, as they are highly effective and safe compared to other standard chemotherapeutic drugs. [32–34] The drug design research field affirms that thiazoles and thiazolidinones are effective moieties in chemotherapeutic drugs. They participate in the construction of many significant compounds with different biological activities. [35–39] There are thiazole-containing compounds that have been reported as potent anticancer agents that target the kinases signaling pathway [40] such as Dasatinib and Dabrafenib (Fig. 1) that considered as thiazole selective drugs with tyrosine kinase inhibitory activity. [41–42] In addition there are approved drugs containing thiazolidinone core structures such as Etozoline and Ralitoline (Fig. 1). Moreover, many thiazolyl-containing compounds have been found to be potent cytotoxic agents against different cell lines. As an example, compound I showed potent cytotoxic activity towards mammary gland breast cancer cell line MCF-7 (acronym for Michigan Cancer Foundations-7) with IC50 of 0.07 µM besides being EGFR inhibitor with IC50 of 0.06 µM. [43, 44] Additionally, the thiazolyl-3-arylpyrazole-4-carbaldehyde II has a significant cytotoxic activity against the cervical epithelioid carcinoma cell line HeLa with IC50 of 5.75 µM. [45] Also, Channar et al. have designed and synthesized thiazolidinone III with excellent cytotoxic activity in MCF-7 and HeLa cell lines. [46] Furthermore, the indole-thiazolidinone IV revealed cytotoxic activity in MCF-7, colon cancer HCT116 and lung cancer A549. [47] Besides, the thiazolone-pyrazoles V-VI represented potent EGFR inhibition capability. [48] In addition, the arylidene derivatives of thiazolidinone VIIa-c showed cytotoxic activity in MCF-7 and hepatocellular carcinoma HepG2. [49] It was alsorecently reported that thiazolidinone VIII exhibited antiproliferative activity targeting HepG2, MCF-7 and HCT116 cell lines (Fig. 2). [50]
Taking into account the aforementioned data and as an extension of our ongoing research work on the designing biologically active heterocyclic products that act anticancer agents and others, [51–64] herein we adopted design and synthesis of new compounds containing thiazolidinone scaffold. Thiazolidinones 3a,b were synthesized from the starting thiosemicarbazones 2a,b, then 3a,b undergoing Knӧvenagel condensation with different aryl and heteryl aldehydes by a two-step or multicomponent route to afford three series of thiazolidinones 5a-j, 7a-h and 9a-j bearing substitution at position-5 (Fig. 3). Twelve of the newly synthesized products were examined for their cytotoxic activity towards MCF-7, HepG2 and HeLa, besides the most potent compounds were evaluated against the normal WI-38 cell line detected their safety character toward normal human cells. Thiazolidinones 5c and 5h, the most potent anticancer candidates were evaluated as target protein kinase inhibitors against EGFR and their effects on the cell cycle and their ability to induce apoptosis in the HepG2 cell line were also investigated. A docking study was also operated to predict the binding modes of the compounds 5c and 5h within the binding site of EGFR using the PDB file (1M17 is the crystal structure of epidermal growth factor receptor with the co-crystallized ligand Erlotinib). Furthermore, in silico toxicity potential by Osiris methodology was performed on twelve synthesized compounds, all of which had zero toxicity risks and were predicted to have good physicochemical properties, thus compounds 5c and 5h were detected to be safe as drug candidates with favorable properties.
2.1. Chemistry
General
The equipment used for detecting the spectral (IR, 1H NMR, 13C NMR and Mass) and physical analyses (melting points, and color) are provided in details in ESI. The elemental analyses and spectral analyses were carried out through the microanalytic center at Cairo University and microanalytic unit-FOPC-NMR Unit-Faculty of Pharmacy-Cairo University. The anticancer investigations were carried out at Faculty of Pharmacy-El Mansoura University. The aldehydes 4-formylphenyl benzoate 1a and 4-formyl-2-methoxyphenyl benzoate 1b were prepared according to the reported method. [65, 66]
Procedure for the synthesis of thiosemicarbazones 2a,b
An equimolar of the aldehydes 1a,b (0.01 mol) and thiosemicarbazide (0.01 mol) was added to a solution of absolute ethanol (10 ml) and glacial acetic acid (3 drops), then the reaction mixture was refluxed for 4 hrs. The reaction mixture was cooled and filtered off then the product was recrystallized from ethanol-dioxane mixture.
Procedure for the synthesis of thiazolidinone derivatives 3a,b
Method A
Aldehydes 2a,b (0.01 mol) was refluxed with choloroacetic acid (0.01 mol) and fused sodium acetate (0.01 mol) in glacial acetic acid (10 ml) for 4 hours. Thiazolidinones 3a,b were collected through filtration, washed by ethanol and then recrystallized from glacial acetic acid.
Method B
Aldehydes 2a,b (0.01 mol) refluxed with ethyl bromoacetate (0.01 mol) in pure ethanol (10 ml) containing fused sodium acetate (0.01 mol) for 4 hours. Thiazolidinone derivatives 3a,b were collected through filtration and recrystallized from glacial acetic acid.
The detailed procedures and the spectral data together with the physical data for all the synthesized compounds were discussed in details in the ESI.
2.2. Biological activity
2.2.1. In Vitro Anticancer screening
MTT assay
It was operated in the Faculty of Pharmacy-El Mansoura University. The Cell lines used are HepG2, MCF-7, and HeLa and they are provided through VACSERA [Holding company for biological products and vaccines]. The MTT assay was conducted according to the literature. [67] The procedure is discussed in details in the ESI.
2.2.2 The inhibitory assay for EGFR kinase
Compounds 5c and 5h, were evaluated for their inhibitory activities against EGFR. The procedure is discussed in details in the ESI
2.2.3. Cell cycle by In vitro DNA-flow cytometric analysis
The first step in the technique is to prepare the HepG2 cells in 25 cm2 cell culture flask. The compounds 5c and 5h were prepared at their IC50 that obtained from MTT assay then treated with RPMI-1640 medium separately. The procedure is discussed in details in the ESI.
2.2.4. The apoptosis assay by Annexin V-FITC analysis
HepG2 cells were harvested and incubated with compound 5c and 5h separately. The experiment was carried by the BioVision Annexin V-FITC Apoptosis Detection Kit. The procedure is discussed in details in the ESI.
2.3. Computational studies
2.3. 1. Molecular docking
The 2D structure of 5c and 5h was drawn through chemdraw program 2021 then each were saved as MDL Molfile (*.mol). The protonated 3D structure of 5c and 5h were applied to measure their bond lengths and angles in their binding pattern with the active site of the co-crystallized structure of EGFR protein kinase with its ligand erlotinib was downloaded (PDB code: 1M17) using MOE program version 2015[Molecular Operating Environment]. The co-crystallized structure of EGFR protein kinase with its ligand erlotinib was downloaded (PDB code: 1M17) from protein data bank ((www.rcsb.org/structure/1m17 ). The procedure is discussed in details in the ESI.
2.3.2. In silico toxicity potential.
Physicochemical characteristics and toxic hazards of 3-d, 5f, 5h, 5i, 7a, 7e and 9a were detected through Osiris methodology [68] and the details were mentioned within the ESI.
3.1. Chemistry
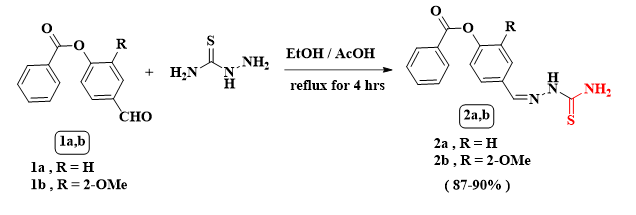
Thiosemicarbazide reacted with each of 4-formylphenyl benzoate 1a and 4-formyl-2-methoxyphenyl benzoate 1b in ethanol containing a catalytic amount of glacial acetic acid afforded the corresponding thiosemicarbazones 2a,b. In compound 2a`s IR spectrum, there are bands at υmax 3455 and 3285 cm− 1 for NH2 and NH functions besides a band for carbonyl group at υmax 1734 cm− 1. The 1H NMR showed a D2O broad signal at δ = 6.51 ppm for NH2`s protons besides another singlet D2O band at δ ~ 10.30 ppm for NH amide group`s proton. Additionally, there are three doublet bands at δ = 7.29, 7.80 and 8.12 ppm with J coupling; 8.7, 8.7 and 7.2 Hz, respectively, all together with another multiple signals at δ = 7.58–7.74 ppm for aromatic protons. Its 13C NMR spectrum displayed significant signals at δ = 151.5, 157.2 and 165.0 ppm for CO, =CH and C = S respectively, with another expected signals at δ = 122.6, 128.2, 129.4, 129.5, 130.3, 133.2, 134.6 and 138.8. (See exp., Scheme 1).
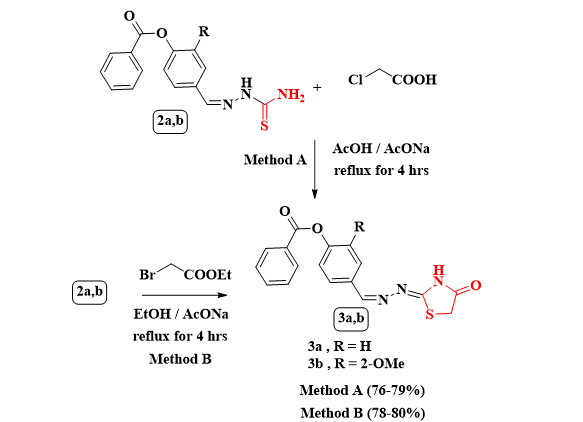
Furthermore, the reaction of the thiosemicarbazones 2a,b with chloroacetic acid in refluxing acetic acid containing equivalent amount of fused sodium acetate afforded the respective thiazolidinones 3a,b (Scheme 2). In the IR spectrum of 3b, presented a broad band at υmax = 3421 cm− 1 for imino group with other bands at υmax = 1727 and 1633 cm− 1 for carbonyl groups. The 1H NMR spectrum of 3b revealed singlet signals at δ = 3.81 and 3.88 ppm for methoxy`s and methylene`s protons respectively, besides another three doublet signals at δ = 7.32, 7.61 and 8.11 ppm with J coupling; 8.1, 7.8 and 7.2 Hz, respectively. Also, the spectrum showed a D2O singlet signal at δ ~ 12.01 ppm for NH`s proton, in addition to another expected signals for aromatic protons and a singlet signal at δ = 8.43 ppm for vinylic proton. Its 13C NMR spectrum displayed significant signals at δ = 164.3, 155.8, 151.6, 56.3 and 33.5 ppm for two CO, C = N of the thiazolidinone ring, OCH3`s and CH2`s carbon respectively. The Mass spectrum of 3b revealed a molecular ion peak at m/z = 369 (M+, 4.55%) which was constituent with the molecular formula C18H15N3O4S (See exp. Scheme 2). The thiazolidinones 3a,b were prepared through another alternative method through the reaction of thiosemicarbazones 2a,b with ethyl bromoacetate and equivalent amount of fused sodium acetate resulted in thiazolidinones 3a,b after 4 hrs under reflux with better yield% [ yield% of 3a; 80 and yield% of 3b; 78 ]. The physical aspects and spectral data for 3a,b obtained by this method were similar as the method A.
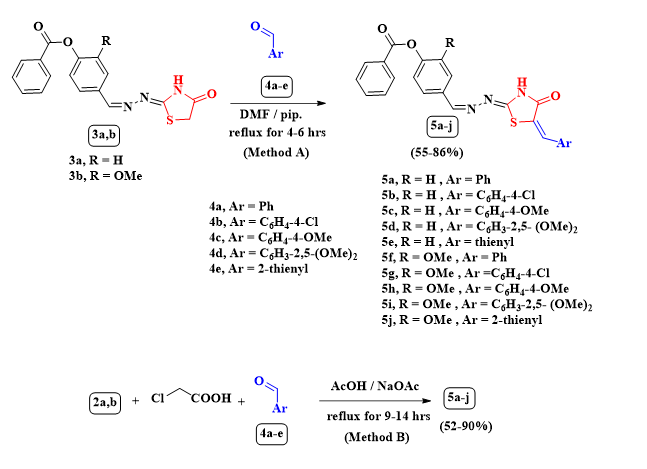
Knӧvenagel condensation of thiazolidinones 3a,b with different aryl or heteryl aldehydes 4a-e in N,N-dimethylformamide concerning few drops of piperidine formed the corresponding arylidenes 5a-j (Scheme 3). In the 1H-NMR spectrum of 5d, the CH2`s protons at δ = 3.91 ppm were absent instead the two OCH3`s protons appeared at δ = 3.80 and 3.85 ppm besides a D2O exchangeable signal at δ = 12.65 ppm for imino proton, also another multiplet signals referred to aryl and vinylic signals at δ = 6.86–7.09 and 7.76–7.80 ppm, respectively, besides three doublet signals at δ = 7.44, 7.92 and 8.15 ppm with J coupling constants 8.0, 8.0 and 7.2 Hz, respectively. The IR spectrum for 5d showed absorption bands at wavelengths 3433, 1738 and 1635 cm− 1 for imino and carbonyl groups. There are significant signals in the 13C NMR spectrum of 5d at δ = 174.9, 164.8, 155.7, 153.4, 55.8 and 56.2 ppm for two CO, two C = N and two C-OCH3 respectively, with another expected signals. The spectral data together with elemental data were in agreement with the suggested structures 5a-j (See exp. Scheme 3). The alternative pathway of the preparation of 5a-j through multicomponent reaction (Method B). So an equimolar amounts of 2a,b, chloroacetic acid and the aldehydes 4a-e in refluxing glacial acetic acid containing fused sodium acetate afforded the same products of the above pathway (Method A) in all physical and spectral aspects (Scheme 3, See Table 1).
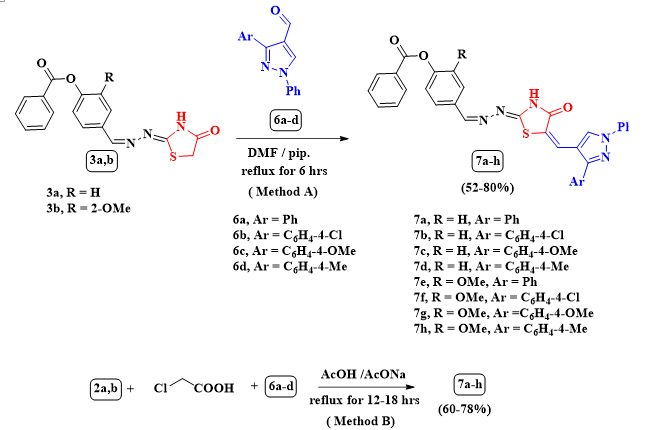
Furthermore, compounds 3a,b were condensed with pyrazole-4-carbaldehyde derivatives 6a-d delivering the respective arylidene derivatives 7a-h according to method A (Scheme 4). The structures of 7a-h were confirmed by spectral tools and elemental analyses, taken for example the spectral data for 7g, its IR spectrum showed a broad band at νmax = 3422 cm-1 for NH group and bands at νmax 1728 and 1648 cm-1 for carbonyl groups. In its 1H-NMR spectrum there are two singlet signals at δ = 3.85 and 3.91 ppm for two methoxy protons and a D2O exchangeable singlet signal at δ = 12.55 ppm due to NH proton, with another expected multiplet signals at δ = 7.34-7.54 and 7.69-7.80 ppm for aryl protons besides doublet signals were found at d = 7.56, 7.60, 7.66, 8.03 and 8.14 ppm with J coupling constants 7.6, 7.6, 7.2, 7.6 and 7.2 Hz, repetitively besides two singlet signals at d = 8.44 and 8.84 ppm for vinylic protons and one singlet signal at d = 8.56 ppm for pyrazole`s proton. Alternatively, the arylidene derivatives 7a-h were obtained through the multicomponent reaction through three components: 2a,b, chloroacetic acid and the pyrazole-4-carbaldehydes 6a-d in refluxing glacial acetic acid and fused sodium acetate (Method B) affording the same products of the above two step pathway similar in the physical and spectral aspects in different yield% (See Table 1, See Exp. Scheme 4).
Furthermore, compounds 3a,b were condensed with pyrazole-4-carbaldehyde derivatives 6a-d delivering the respective arylidene derivatives 7a-h according to method A (Scheme 4). The structures of 7a-h were confirmed by spectral tools and elemental analyses, taken for example the spectral data for 7g, its IR spectrum showed a broad band at νmax = 3422 cm− 1 for NH group and bands at νmax 1728 and 1648 cm− 1 for carbonyl groups. In its 1H-NMR spectrum there are two singlet signals at δ = 3.85 and 3.91 ppm for two methoxy protons and a D2O exchangeable singlet signal at δ = 12.55 ppm due to NH proton, with another expected multiplet signals at δ = 7.34–7.54 and 7.69–7.80 ppm for aryl protons besides doublet signals were found at δ = 7.56, 7.60, 7.66, 8.03 and 8.14 ppm with J coupling constants 7.6, 7.6, 7.2, 7.6 and 7.2 Hz, repetitively besides two singlet signals at δ = 8.44 and 8.84 ppm for vinylic protons and one singlet signal at δ = 8.56 ppm for pyrazole`s proton. Alternatively, the arylidene derivatives 7a-h were obtained through the multicomponent reaction through three components: 2a,b, chloroacetic acid and the pyrazole-4-carbaldehydes 6a-d in refluxing glacial acetic acid and fused sodium acetate (Method B) affording the same products of the above two step pathway similar in the physical and spectral aspects in different yield% (See Table 1, See Exp. Scheme 4).
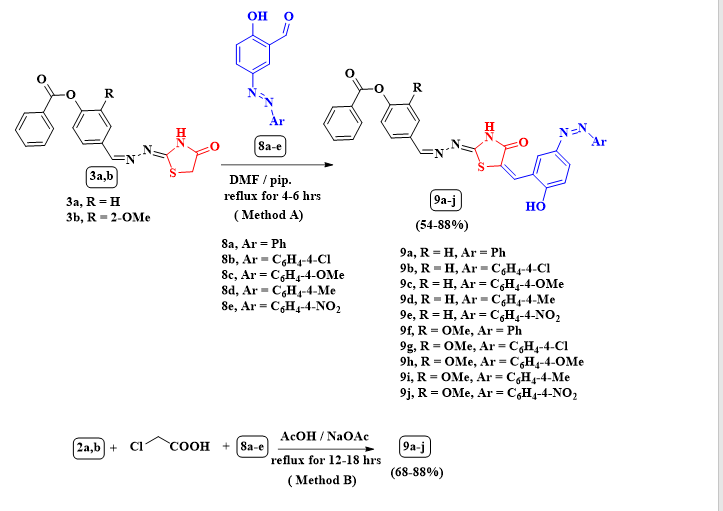
Finally, under the same reaction conditions of method A, the condensation of compounds 3a,b with 5-arylazo-2-hydroxybenzaldehydes 8a-e afforded the respective arylidenes 9a-j (Scheme 5). The IR spectrum of 9c exposed absorption bands appeared at υmax 3412 and 3242 cm− 1 attributed to imino and hydroxyl functions, also absorption bands at υmax 1736 and 1637 cm− 1 attributed to CO functions, respectively. In the 1H NMR spectrum of 9c there are five doublet signals at δ 6.81, 7.13, 7.47, 7.92 and 8.13 ppm for aryl protons with J coupling constants = 8.1, 8.4, 8.1, 8.1 and 8.4 Hz and a multiplet signal at 7.30–7.44 ppm and 7.52–7.88 ppm for phenyl protons, and two exchangeable D2O signals at = 9.97 and 11.91 ppm due to imino and hydroxyl protons. Additionally, there were two singlet signals at δ 8.26 and 8.56 ppm due to vinylic protons. The products 9a-j were obtained also alternatively through one-pot reaction of 2a,b, chloroacetic acid and arylazo salicyladehydes 8a-e (Method B) (See Table 1, Scheme 5).
Table 1 Yield% of 5a-j, 7a-h and 9a-j obtained by Method A and Method B revealing the reaction time in each method
|
Compound No. |
Method A |
Method B |
||
|---|---|---|---|---|
|
Yield% |
Time (hrs) |
Yield% |
Time (hrs) |
|
|
5a |
70 |
4 |
90 |
9 |
|
5b |
78 |
4 |
70 |
9 |
|
5c |
78 |
4 |
78 |
10 |
|
5d |
78 |
5 |
76 |
12 |
|
5e |
55 |
6 |
52 |
14 |
|
5f |
86 |
4 |
80 |
14 |
|
5g |
74 |
5 |
72 |
12 |
|
5h |
55 |
5 |
58 |
12 |
|
5i |
64 |
6 |
60 |
12 |
|
5j |
78 |
6 |
72 |
14 |
|
7a |
75 |
6 |
78 |
12 |
|
7b |
78 |
6 |
75 |
12 |
|
7c |
78 |
6 |
76 |
15 |
|
7d |
78 |
6 |
70 |
14 |
|
7e |
52 |
6 |
60 |
14 |
|
7f |
78 |
6 |
68 |
14 |
|
7g |
68 |
6 |
68 |
13 |
|
7h |
80 |
6 |
76 |
18 |
|
9a |
78 |
4 |
80 |
12 |
|
9b |
73 |
4 |
78 |
12 |
|
9c |
80 |
5 |
78 |
12 |
|
9d |
52 |
6 |
68 |
16 |
|
9e |
78 |
6 |
88 |
16 |
|
9f |
64 |
6 |
70 |
14 |
|
9g |
56 |
6 |
68 |
14 |
|
9h |
54 |
6 |
70 |
18 |
|
9i |
88 |
6 |
82 |
18 |
|
9j |
58 |
6 |
74 |
18 |
3.2. Biological activities
3.2.1. Cytotoxic activity assessment by in vitro MTT assay
The tested compounds` cytotoxicity and anticancer activity against MCF-7, HepG2 and HeLa was detailed in Table 2. The MTT assay was used for screening the anticancer activity of the tested compounds. [67] Furthermore, these compounds were examined against normal lung fibroblast (WI38) to find if they are safe towards the normal cells. Sorafenib was used as a reference drug and the obtained data for the cytotoxicity of the compounds in the three cell lines (IC50, µM) was detailed in Table 2. According to the revealed data the compounds under test displayed versatile anticancer activity toward the three cell lines showing moderate to very strong activity. The starting thiazolidinones 3a and 3b showed strong activity in MCF-7, HepG2 and HeLa with IC50 values for 3a; 17.26 ± 1.5, 14.50 ± 1.1 and 11.66 ± 0.9 µM, respectively, while IC50 for 3b; 17.71 ± 1.4 and 21.26 ± 1.5 µM in MCF-7 and HepG2, respectively. Compound 3b exhibited weak anticancer activity in HeLa with IC50 = 50.67 ± 2.8 µM. The compound 5c showed high potency in HepG2 and HeLa with IC50; 9.15 ± 0.6 and 9.18 ± 0.7 µM, respectively. Compared with sorafenib, 5c was more potent in HepG2 with IC50 which was in an equipotent manner to sorafenib (IC50; 9.18 ± 0.6 µM). The compound 5h produced the most significant cytotoxic activity towards the cell lines under study among all the tested compounds. It showed more potency than the standard in HepG2 and HeLa with IC50 values; 6.22 ± 0.4 and 9.18 ± 0.7 µM, respectively, comparing with sorafenib with IC50 values; 9.18 ± 0.6 and 8.04 ± 0.5 µM, respectively. The presence of 4-methoxyphenyl moiety in the thiazolidinone ring of 5c and 5h produced enhancement in the anticancer activity.
The most active compounds 3a, 3b, 5a, 5c and 5h were examined for their cytotoxicity against the normal fibroblasts (WI38) cell line. The tested compounds showed higher IC50 towards WI38 cell lines as they had cytotoxicity with IC50; 80.07 ± 3.9, 83.87 ± 4.1, 38.20 ± 2.4, 79.30 ± 3.7 and 57.54 ± 3.2 µM for 3a, 3b, 5a, 5c and 5h, respectively. From these results, it was concluded that 3a, 3b, 5a, 5c and 5h can be used as anticipated anticancer agents targeting only the cancerous cells (Table 2, Fig. 4).
|
3a |
14.50 ± 1.1 |
17.26 ± 1.5 |
11.66 ± 0.9 |
80.07 ± 3.9 |
|---|---|---|---|---|
|
Sorafenib |
9.18 ± 0.6 |
7.26 ± 0.3 |
8.04 ± 0.5 |
10.65 ± 0.8 |
|
Compound NO. |
Cell line (HepG2) IC50 (µM) |
Cell line (MCF-7) IC50 (µM) |
Cell line (HeLa) IC50 (µM) |
Cell line (WI38) IC50 (µM) |
|
3b |
21.26 ± 1.5 |
17.71 ± 1.4 |
50.67 ± 2.8 |
83.87 ± 4.1 |
|
5a |
26.98 ± 1.9 |
20.73 ± 1.8 |
19.07 ± 1.4 |
38.20 ± 2.4 |
|
5b |
31.47 ± 2.1 |
25.50 ± 2.0 |
86.01 ± 4.0 |
|
|
5c |
9.15 ± 0.6 |
13.55 ± 1.1 |
9.18 ± 0.7 |
79.30 ± 3.7 |
|
5d |
43.18 ± 2.6 |
35.08 ± 2.3 |
36.59 ± 2.2 |
|
|
5f |
98.90 ± 4.4 |
51.98 ± 2.9 |
74.30 ± 3.6 |
|
|
5h |
6.22 ± 0.4 |
9.39 ± 0.7 |
7.78 ± 0.5 |
57.54 ± 3.2 |
|
5i |
52.27 ± 2.9 |
36.24 ± 2.4 |
30.89 ± 2.1 |
|
|
7a |
78.65 ± 3.6 |
45.15 ± 2.7 |
59.98 ± 3.3 |
|
|
7e |
108.60 ± 4.8 |
63.96 ± 3.3 |
64.05 ± 3.4 |
|
|
9a |
63.86 ± 3.2 |
41.38 ± 2.5 |
23.79 ± 1.9 |
|
|
IC50: Compound concentration required to cause cell death by 50%, IC50 values = mean ± SD |
||||
3.2.2. The Inhibitory activity of compounds 5c and 5h towards EGFR tyrosine kinase.
The most cytotoxic products 5c and 5h were examined as target protein kinase inhibitors for EGFR tyrosine kinase and erlotinib was used as standard drug. [69, 70] The obtained data was summarized in Table 3. Compound 5c showed decrease in the inhibitory activity against EGFR kinase protein with IC50 value; 0.2 µM compared to the reference drug erlotinib which had IC50 value; 0.037 µM. While, the thiazolidinone derivative 5h appeared to be more potent inhibitor against EGFR kinase activity giving IC50 value; 0.098 µM. It could be noticed that the presence of donating substitution in the pharmacophore of 5c and 5h caused a positive effect in the inhibition against EGFR kinase and the increase in the number of that kind of substitution in turn increased the inhibitory effect as seen in 5h. [71]

3.2.3. Cell cycle analysis for 5c and 5h by DNA-flow cytometric assay
Moreover, the compounds 5c and 5h were evaluated for their effect on the cell cycle in HepG2 cell lines. The stages of cell cycle were detected through flow cytometry after propidium iodide (PI) staining [69]. From the obtained results, it was concluded that 5c and 5h caused interruption in the cell cycle progression in the treated HepG2 cells. The change in the cell content % was detected in the treated cells with 5c and 5h compared to the control cells. It was observed that the content of the cells at pre-G1 stage increased in the treated HepG2 cells from 44.41–52.48% a decrease in the quantity of cells at S phase and G2/M phase from in 35.49% and 19.7% (Fig. 5, B) so that it was clearly detected that 5c arrested the cell cycle in G1 phase. While treatment of HepG2 cells with 5h, there was an increase in the quantity of cells was observed at pre-G1 and S phases to reach 47.22% and 42.04%, respectively causing a remarkable decrease in the cell content in the G2/M phase to reach 10.74% in treated cells, for that observation, it was clear that 5harrested the cell cycle at the late G1 phase and the beginning of S phase (Fig. 5, C).
3.2.4. Determination of apoptosis induction for 5c and 5h by annexin V-FTTC method
The cell death of HepG2 cell line by apoptotic pathway induced by compound 5c and 5h was determined by annexin V/PI assay [69]. Annexin V-FITC stain was used to stain cells with PI dye. The principle of this method is the cells that are in the late apoptosis stage are stained positive with V/PI. The HepG2 cells were treated with compounds 5c and 5h at their IC50 concentrations 9.15 ± 0.6 and 6.22 ± 0.4 µM, respectively for 24 hr after that the cells were stained by annexin V / PI and then the corresponding red PI and green FITC fluorescence was detected by flow cytometry technique. The representative dot plots for compounds 5c and 5h of flow cytometric analyses showing four various distributions (Fig. 6). After treatment with 5c, 11.93% of the cells were in early apoptosis and 21.95% were in a late apoptosis phase, respectively (Table 5, Fig. 6, B). While cells treated with 5h revealed that 28.51% of the cells were in early apoptosis and 16.36% were in a late apoptosis phase, respectively (Fig. 6, C). As concluded, the cell death of the treated HepG2 cells was caused mainly by apoptosis after treatment with compounds 5c and 5has low content of these cells was in necrosis stage.


3.3. Computational studies
3.3.1. Molecular modeling and Docking Study on EGFR
The anticancer investigated compounds 5c and 5h were subjected for Molecular docking using Molecular operating environment [MOE dock 2015] software. The docking studies were subjected to determine the possible binding pattern between the 3D structure of 5c and 5h with the active site pocket of EGFR kinase. The self-docking of the active site of EGFR with the co-crystallized ligand erlotinib (PDB code: 1M17) has an energy score of -7.8094 Kcal mol− 1 and RMSD value of 0.9361Å between erlotinib and its docked pose. As depicted in Fig. 7, the compound 5c was bonded to the active site of EGFR with energy score − 11.9196 Kcal mol− 1 and RMDS value 1.3455 Å with the formation of hydrogen-arene interaction through the thiazolidinone centroid and the amino acid Gly 772 with bond length equal 3.67 Å.
By inspection of Fig. 8, the derivative 5h was bonded to the vicinity of EGFR` active site with the formation of one hydrogen bond acceptor between the oxygen atom of carbonyl group and the backbone`s amino acid Thr 830 with bond length value 2.59 Å. The energy score of the interaction was − 12.1942 Kcal mol− 1 and the RMDS value 1.7160 Å.
Finally, regarding to the superimposition Fig. 7A and 8A, it was noted that the presence of thiazolidinone ring in compound 5c and the presence of carbonyl group in 5h gave a chance for better insertion of these compounds into the active site of EGFR with similar bonding pattern as the co-crystallized ligand erlotinib through hydrogen-arene and hydrogen bond formations.
3.3.1. In silico toxicity potential by Osiris property explorer
The newly synthesized compounds 3a, 3b, 5a, 5b, 5c, 5d, 5f, 5h, 5i, 7a, 7e and 9a were examined for detecting their toxic effects such as mutagenic, tumorigenic properties and skin irritants besides predicting their physicochemical properties via Osiris methodology, [68] and the obtained results were detailed in Table 6. From the obtained results, it was concluded that all the compounds under study have no tendency to be mutagenic, tumorigenic or even cause skin irritation and have no reproductive effect except compound 9a which showed to have toxicity risks. The compounds 3a and 3b showed to have a better drug score of values; 0.61 and 0.6 respectively with better TPSA values that are \(\le\)140 Å (3a has TPSA = 105.4 Å, 3b has TPSA = 114.6 Å). Compound 5a has drug score = 0.3 and TPSA value; 105.4 Å. The most cytotoxic compounds 5c and 5h showed to have almost the same drug score of values; 0.29 and 0.28 respectively with TPSA values; 114.6 Å and 123.8 Å, respectively. The compounds 5c and 5hshowed their effectiveness and potentiality as new drugs.
|
Comp. no. |
Mutagenicity |
Tumorgenicity |
Irritancy |
Reproductive effect |
Solubility |
Drug-likeness |
Drug score |
TPSA |
|---|---|---|---|---|---|---|---|---|
|
Toxicity risks |
||||||||
|
3a |
Green |
Green |
Green |
Green |
-4.98 |
4.35 |
0.61 |
105.4 |
|
3b |
Green |
Green |
Green |
Green |
-5.0 |
5.59 |
0.6 |
114.6 |
|
5a |
Green |
Green |
Green |
Green |
-6.81 |
4.35 |
0.3 |
105.4 |
|
5b |
Green |
Green |
Green |
Green |
-7.55 |
6.04 |
0.25 |
105.4 |
|
5c |
Green |
Green |
Green |
Green |
-6.83 |
5.43 |
0.29 |
114.6 |
|
5d |
Green |
Green |
Green |
Green |
-6.85 |
6.05 |
0.28 |
123.8 |
|
5f |
Green |
Green |
Green |
Green |
-6.83 |
5.53 |
0.29 |
114.6 |
|
5h |
Green |
Green |
Green |
Green |
-6.85 |
6.82 |
0.28 |
123.8 |
|
5i |
Green |
Green |
Green |
Green |
-6.86 |
6.85 |
0.26 |
133.1 |
|
7a |
Green |
Green |
Green |
Green |
-8.32 |
7.52 |
0.19 |
123.2 |
|
7e |
Green |
Green |
Green |
Green |
-8.34 |
8.59 |
0.18 |
132.4 |
|
9a |
Red |
Red |
Orange |
Red |
-9.01 |
-2.94 |
0.02 |
150.3 |
|
Green color: shows less toxic, Orange color: shows mid toxic, Red color: shows high tendency of toxicity |
||||||||
This research detailed synthesis of three series of thiazolidinone derivatives based on 4-formylphenyl benzoate 1a and 4-formyl-2-methoxyphenyl benzoate 1b as key precursors and most of these compounds were subjected for MTT assay for investigating their cytotoxicity towards three human cancer cell lines; hepatocellular HepG2, breast MCF-7 and cervical HeLa, using sorafenib as a reference standard. The compounds 5c and 5h revealed better inhibitory activities towards the three cancer cell lines with IC50 values for 5c; 9.15, 13.55 and 9.18 μM in HepG2, MCF-7 and HeLa, respectively and IC50 values for 5h; 6.22, 9.39 and 7.78 μM in HepG2, MCF-7 and HeLa, respectively, comparing to sorafenib of IC50; 9.18, 7.26 and 8.04 μM in HepG2, MCF-7 and HeLa, respectively. Moreover, compounds 5c and 5h revealed an excellent safety pattern against the normal WI38 cell line. Additionally, compounds 5c and 5h were examined as targeting protein kinase inhibitors against EGFR, the compound 5c showed inhibitory suppression effect of IC50; 0.2 μM while 5h showed potent inhibition impact of IC50; 0.098 μM, comparing to the standard erlotinib of IC50; 0.037 μM. Additionally, both 5c and 5h were investigated for their effect on cell cycle progression and induction of cell death by apoptosis in treated HepG2 cells and it was observed their ability in interrupting the cell cycle progression as 5c arrested it in G1 phase and 5h arrested it in the end of G1 phase and the start of S phase and all together caused cell death by apoptosis mechanism. Molecular docking study declared good fitting patterns with good binding energy in the active site of EGFR kinase protein [PDB file: 1M17], in addition toxicity study using Osiris property explorer revealed excellent drug like properties and no toxicity or hazards in humans.
Competing interests
The authors declare no conflict of interest.
Authors' contributions
Nadia Hanafy Metally has generally supervised the work and has provides the conceptions, follow the data interpretations, original manuscript writing and reviewing process handling.
Salwa Magdy Eldaly has provides the conceptions, follow the analyses, data interpretations, original manuscript writing
Dalia salama has performed the experimental work, carried out the needed analysis and data presentation.
Availability of data and materials
The supporting information contains detailed experimental procedures, physical properties [Color, Yield%, m.p], spectroscopic data [IR, 1H NMR, 13C NMR and MS], elemental analyses of the compounds are found in the Supporting Information together with 1H NMR spectra, 13C NMR spectra, IR and MS spectra of some compounds are found.
- A. M. Soliman, A. S. Alqahtani, M. M. Ghorab, Journal of Enzyme Inhibition and Medicinal Chemistry. 34, 1030 (2019).
- J. Ferlay, M. Laversanne, M. Ervik, F. Lam, M. Colombet, L. Mery, M. Pineros, A. Znaor, I. Soerjomataram, F. Bray, Global Cancer Observatory: Cancer Tomorrow, Lyon, France. (International Agency for Research on Cancer, 2020), https://gco.iarc.fr/tomorrow.
- S. O. Zaraei, R. M. Sbenati, N. N. Alach, H. S. Anbar, R. ElGamal, H. Tarazi, M. K. Shehata, M. S. Abdel-Maksoud, C.-H. Oh, M. I. El-Gamal, Eur. J. Med. Chem. 224, 113674 (2021).
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal, F. Bray. CA Cancer J Clin.71, 209 (2021).
- International Agency for Research on Cancer. (GLOBOCAN IARC 12 sept 2018), https://www.uicc.org/news/global-cancer-data-globocan-2018.
- R. Awasthi, A. Roseblade, P. M. Hansbro, M. J. Rathbone, K. Dua, M. Bebawy, Curr. Drug Targets. 19, 1696 (2018).
- V.V. Padma, Biomedicines. 5, 1 (2015).
- Z. Du, C. M. Lovly, Mol. Cancer. 17, 58 (2018).
- N. Iqbal, N. Iqbal, Chemother. Res. Pract. 2014, 357027 (2014).
- B. J. Druker, Trends Mol. Med. 8, S14 (2002).
- K. S. Bhullar, N. O. Lagar´on, E. M. McGowan, I. Parmar, A. Jha, B. P. Hubbard, H. V. Rupasinghe, Mol. Cancer. 17, 1 (2018).
- H. Kittler, P. Tschandl, Br. J. Dermatol. 178, 26 (2018).
- A. Ayati, S. Moghimi, S. Salarinejad, M.Safavi, B. Pouramiri, A.Foroumadi, Bioorg. Chem. 99, 103811 (2020).
- E.-M. Birkman, A.Ålgars, M. Lintunen, R. Ristamaki, J. Sundstrom, O.Carpén, BMC Cancer. 16, 406 (2016).
- M. D. Siegelin, A. C. Borczuk, Laboratory Investigation. 94, 129 (2014).
- C. B. Williams, K. Phelps-Polirer, I. P. Dingle, C. J. Williams, M. J. Rhett, S. T. Eblen, K. Armeson, E. G. Hill, E. S. Yeh, Oncogene. 39, 1112 (2020).
- X. Yang, W. Wang, C. Wang, L. Wang, M. Yang, M. Qi, H. Su, X. Sun, Z. Liu, J. Zhang, X. Qin, B. Han, Oncology Reports. 32, 700 (2014).
- N. Normanno, M. R. Maiello, A. De Luca, J. Cell Physiol. 194, 13 (2003).
- Y. Ito, T. Takeda, M. Sakon, M. Tsujimoto, S. Higashiyama, K. Noda, E. Miyoshi, M. Monden, N. Matsuura, Br J Cancer. 84, 1377 (2001).
- S. Kira, T. Nakanishi, S. Suemori, M. Kitamoto, Y. Watanabe, G. Kajiyama, Liver. 17, 177 (1997).
- G. Giannelli, A. Azzariti, C. Sgarra, L. Porcelli, S. Antonaci, A. Paradiso, Biochem Pharmacol. 71 (4), 479 (2006).
- M. Hopfner, A. P. Sutter, A. Huether, D. Schuppan, M. Zeitz, H. Scherubl, J Hepatol. 41, 1008 (2004).
- A. Huether, M. Höpfner, V. Baradari, D. Schuppan, H. Scherübl, Biochem Pharmacol. 70, 1568 (2005).
- P. A. Philip, M. R. Mahoney, C. Allmer, J. Thomas, H. C. Pitot, G. Kim, R. C. Donehower, T. Fitch, J. Picus, C. Erlichman, J Clin Oncol. 23, 6657 (2005).
- R. Kannangai, F. Sahin, M. S. Torbenson, Modern Pathology. 19, 1456 (2006).
- M. R. V. Finlay, R. A. Ward, the Topics in Medicinal Chemistry, (Springer, 28, 2017), pp. 39.
- S. O. Lim, C. W. Li, W. Xia, H. H. Lee, S. S. Chang, J. Shen, J. L. Hsu, D. Raftery, D. Djukovic, H. Gu, W. C. Chang, H. L. Wang, M. L. Chen, L. Huo, C. H. Chen, Y. Wu, A. Sahin, S. M. Hanash, G. N. Hortobagyi, M. C. Hung, Cancer Res. 76, 1284 (2016).
- H.-Y. Lin, W.-X. Sun, C.-S. Zheng, H.-W. Han, X. Wang, Y.-H. Zhang, H.-Y. Qiu, C.-Y. Tang, J.-L. Qi, G.-H. Lu, R.-W.Yang, X.-M. Wang, Y.-H. Yang, RSC Adv. 7, 48404 (2017).
- Y. Yarden, G. Pines, Nat Rev Cancer. 12, 553 (2012).
- R. R. Khattab, A. A. Hassan, D. A. A. Osman, F. M. AbdelMegeid, H. M. Awad, E. S. Nossier, W. A. El-Sayed, Nucleosides, Nucleotides Nucleic acids. 40, 1090 (2021).
- F. Ciardiello, G. Tortora, Clin. Cancer Res. 7, 2958 (2001).
- A. Ayati, S. Moghimi, S. Salarinejad, M. Safavi, B. Pouramiri, A. Foroumadi, Bioorg. Chem. 99, 103811 (2020).
- R. R. Khattab, A. K. Alshamari, A. A. Hassan, H. H. Elganzory, W. A. El-Sayed, H. M. Awad, E. S. Nossier, N. A. Hassan, J. Enzyme Inhib. Med. Chem. 36, 504 (2021).
- I. M. Othman, Z. M. Alamshany, N. Y. Tashkandi, M. A. Gad-Elkareem, M. M. Anwar, E. S. Nossier, Bioorg. Chem. 114, 105078 (2021).
- N. Ahangar, A. Ayati, E. Alipour, A. Pashapour, A. Foroumadi, S. Emami, Chemical Biology & Drug Design. 78, 844 (2011).
- A. Ayati, S. Emami, A. Asadipour, A. Shafiee, A. Foroumadi, European Journal of Medicinal Chemistry. 97, 699 (2015).
- M. Djukic, M. Fesatidou, I. Xenikakis, A. Geronikaki, V. T. Angelova, V. Savic, M. Pasic, B. Krilovic, D. Djukic, B. Gobeljic, M. Pavlica, A. Djuric, I. Stanojevic, D. Vojvodic, L. Saso, Chemico-Biological Interactions. 286, 119 (2018).
- N. B. Patel, H. R. Patel, F. M. Shaikh, D. Rajani, Medicinal Chemistry Research. 23, 1360 (2014).
- K. A. Szychowski, M. L. Leja, D. V. Kaminskyy, U. E. Binduga, O. R. Pinyazhko, R. B. Lesyk, J. Gminski, Chemico-Biological Interactions. 262, 46 (2017).
- A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, Expert Opinion on Therapeutic Patents. 24, 759 (2014).
- G. M. Keating, Drugs. 77, 85 (2017).
- A. Puszkiel, G. Noé, A. Bellesoeur, N. Kramkimel, M.-N. Paludetto, A. Thomas-Schoemann, M. Vidal, F. Goldwasser, E. Chatelut, B. Blanchet, Clinical Pharmacokinetics. 58, 451 (2019).
- P. C. Lv, H. Q. Li, J. Sun, Y. Zhou, H. L. Zhu, Bioorg. Med. Chem. 18, 4606 (2010).
- P. C. Lv, D. D. Li, Q. S. Li, X. Lu, Z. P. Xiao, H. L. Zhu, Bioorg. Med. Chem. Lett. 21, 5374 (2011).
- K. Vaarla, R. K. Kesharwani, K. Santosh, R. R. Vedula, S. Kotamraju, M. K. Toopurani, Bioorg. Med. Chem. Lett. 25, 5797 (2015).
- P. A. Channara, M. Azizb, S. A. Ejazb, G.-e-S. Chaudhryc, A. Saeedb, R. Ujand, A. Hasana, S. R. Ejaze, A. Saeed, JOURNAL OF BIOMOLECULAR STRUCTURE AND DYNAMICS. (2021) https://doi.org/10.1080/07391102.2021.2018045
- A. Kryshchyshyn-Dylevych, L. Radko, N. Finiuk, M. Garazd, N. Kashchak, A. Posyniak, K. Niemczuk, R. Stoika, R. Lesyk, Bioorganic & Medicinal Chemistry. 50, 116453 (2021).
- I. M. M. Othman, Z. M. Alamshany, N. Y. Tashkandi, M. A. M. Gad-Elkareem, S. S. Abd El-Karim, E. S. Nossier, RSC Adv. 12, 561 (2022).
- N. A. Khalil , E. M. Ahmed , H. B. El-Nassan, Med Chem Res. 22, 1021 (2013).
- K. El-Adl, H. Sakr, M. Nasser, M. Alswah, F. M.A. Shoman, Arch. Pharm. 353, e2000079 (2020).
- N. H. Metwally, M. A. Abdallah, M. A. Mosselhi, E. A. El-Desoky, Carbohydrate Research. 345, 1135 (2010).
- N. H. Metwally, M. A. Badway, D. S. Okpy, Chem Pharm Bull. 63, 495 (2015).
- N. H. Metwally, F. M. Abdelrazek, S. M. Eldaly, Res Chem Intemed. 42, 1071 (2016).
- N. H. Metwally, E. A. Deeb, Bioorg. Chem. 77, 203 (2018).
- N. H. Metwally, F. M. Abdelrazek, S. M. Eldaly, J. Heter. Chem. 45, 2668 (2018).
- N. H. Metwally, I. T. Radwan, W. S. El-Serwy, M. A. Mohamed, Bioorg. Chem. 84, 456 (2019).
- N. H. Metwally, M. S. Mohamed, E. A. Ragab, Bioorg. Chem. 88, 102929 (2019).
- N. H. Metwally, G. R. Saad, E. A. Abdwahab Int. J. Nanomed. 20, 6645 (2019).
- N. H. Metwally, F. M. Abdelrazek, S. M. Eldaly, J. Heter. Chem. 57, 3653 (2020).
- N. H. Metwally, S. O. Abdallah, M. M. Mohsen, Biorg. Chem. 97, 103672 (2020).
- N. H. Metwally, M. S. Mohamed,E. A. Deeb, Res. on Chem. Intermed. 47, 5027 (2021).
- N. H. Metwally, A. S. Abd-Elmoety, J. Molecular Str. 1257, 132590 (2022).
- N. H. Metwally, M. A. Badawy, D. S. Okpy, J. Mol. Str. 1258, 132848 (2022).
- N. H. Metwally, S. M. Eldaly, Chemistry Select (2022) https: //doi.org/ 10.1002/ slct.202202257.
- Current Patent Assignee: ROKA FURADADA - WO2011/45389, 2011, A1.
- N. H. Metwally, M. A. Badway, D. S. Okpy, Synth. Commun. 48, 2496 (2018).
- T. Mosmann. J. Immunol. Methods. 65, 55 (1983).
- The OSIRIS property explorer software. Available from: http://www.organic-chem istry.org/prog/peo/.
- M. S. Nafie, S. M. Kishk, S. Mahgoub, A. M. Amer, Chem. Biol. Drug Des. 99, 547 (2022).
- M. W. Aziz, A. M. Kamal, K. O. Mohamed, A. A. Elgendy, Bioorg. Med. Chem. Lett. 41, 127987 (2021).
- O. M. Soltan, M. E. Shoman, S. A. Abdel-Aziz, K. Nagaoka, A. Narumi, M. Abdel-Aziz, J. Adv. Biomed. & Pharm. Sci. 4, 152 (2021).
Schemes are available in the Supplementary Files section.
No competing interests reported.
- supplimentryfile.docx
- Scheme1.png
Scheme 1 Synthesis of thiosemicarbazones 2a,b through the reaction of 1a,b with thiosemicarbazide
- Scheme2.png
Scheme 2 Synthesis of thiazolidinones 3a,b through method A and method B
- Scheme3.png
Scheme 3 Descriptive scheme for the synthesis of 5a-jthrough method A and method B
- Scheme4.png
Scheme 4 Synthesis of 7a-h through method Aand method B
- Scheme5.png
Scheme 5 The reaction of thiazolidinones 3a,b with azo derivatives of salicylaldehydes 8a-e (Method A) and the reaction of 2a,b, chloroacetic acid and 8a-e (Method B) both afforded 9a-j.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}