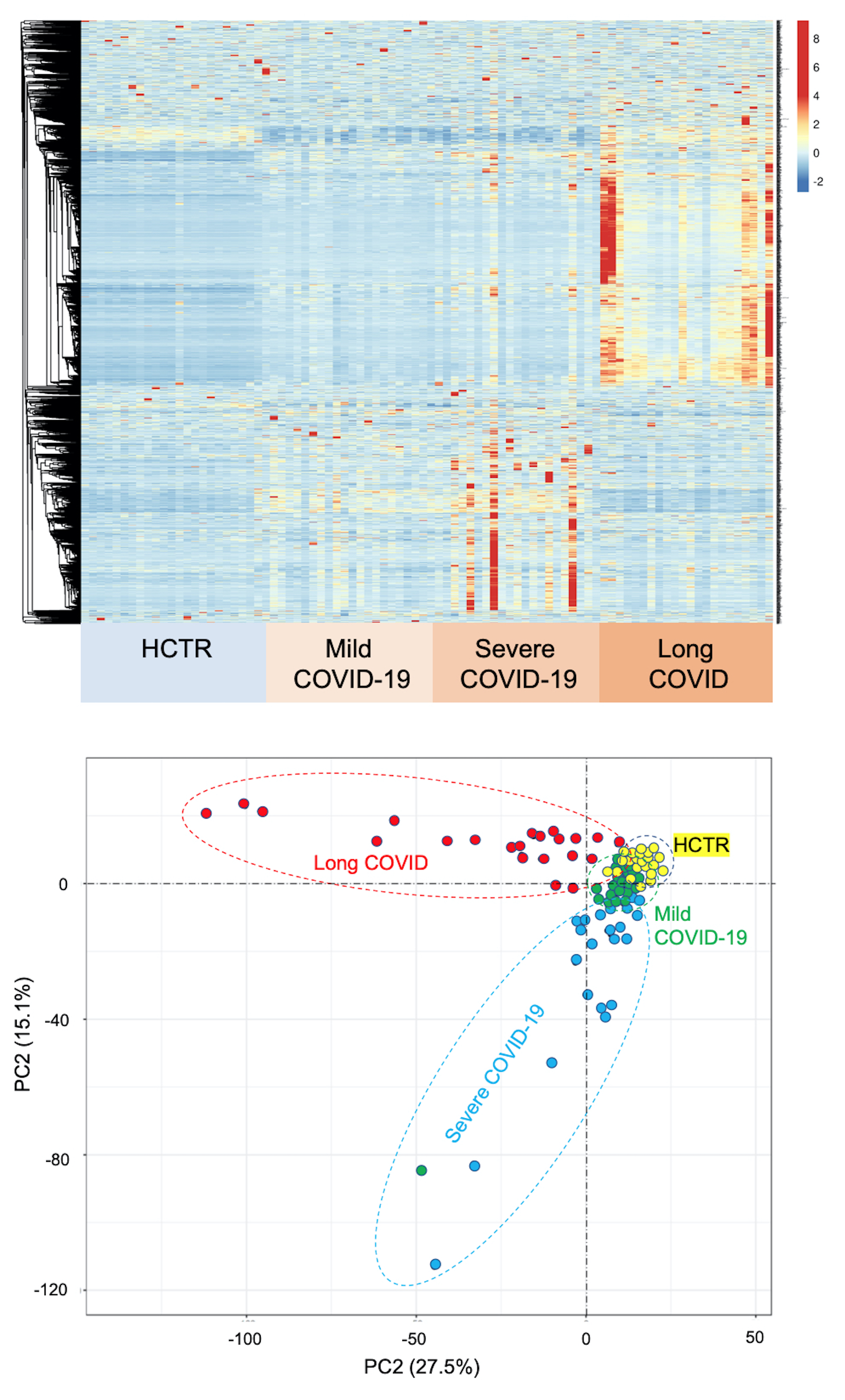
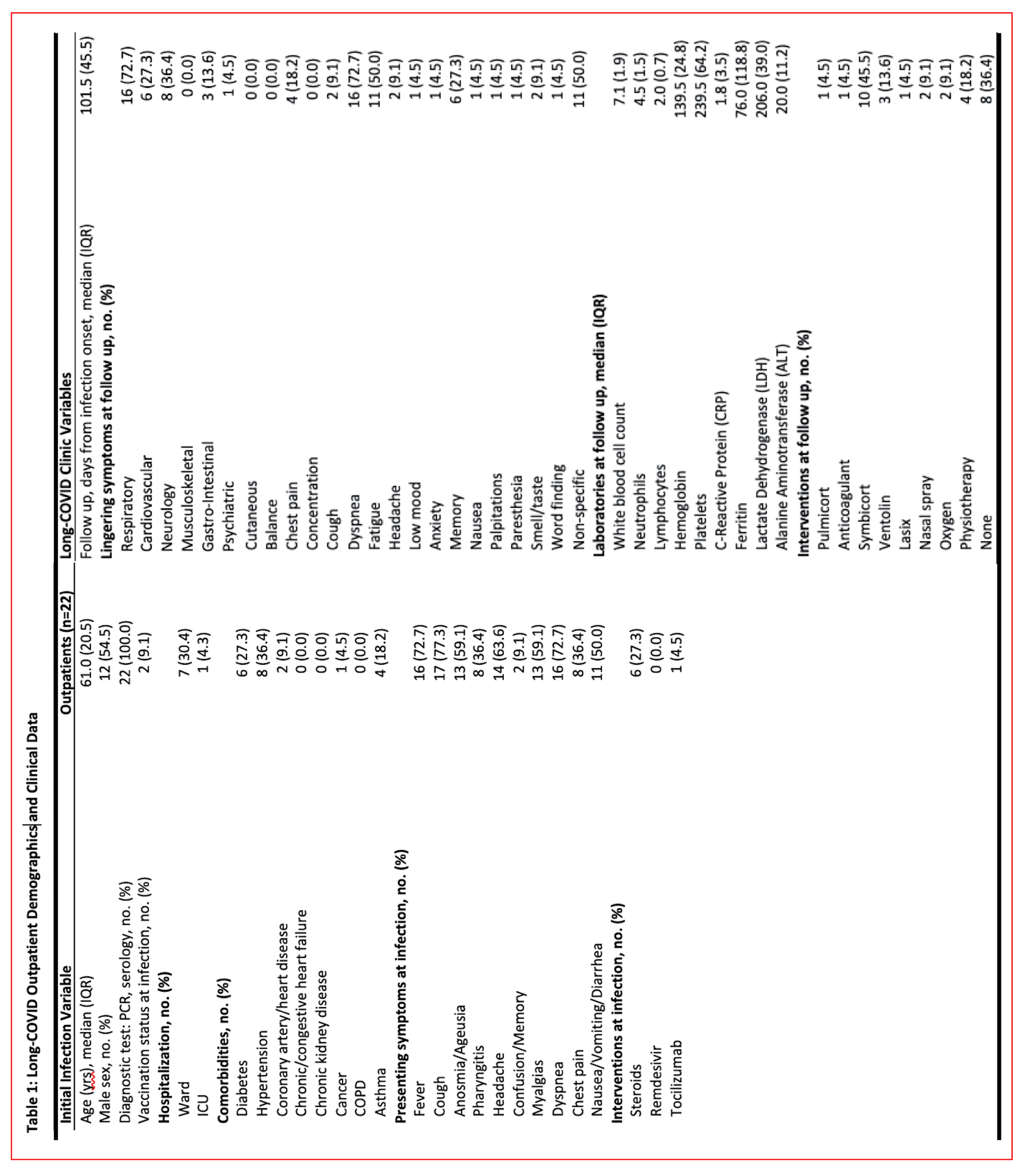
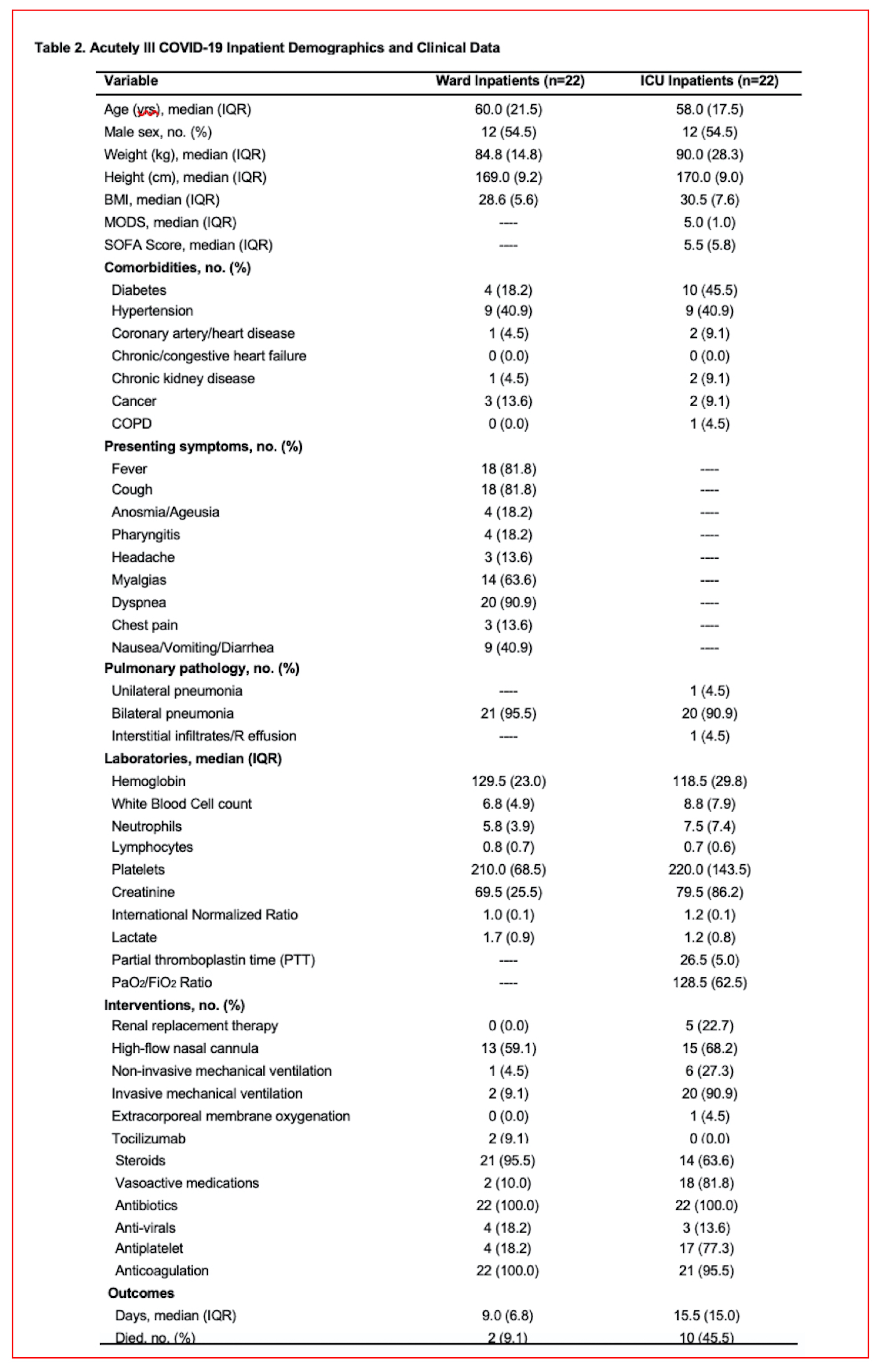
Study model and dimensionality reduction of the data sets. The patient demographic and clinical data are shown in Supplementary Tables 1 (Long-COVID outpatients) and 2 (COVID-19 inpatients). Figure 1A (left panel) illustrates the experimental model consisting of four cohorts of patients, Long-COVID outpatients, acutely ill COVID-19 inpatients (evere), mild COVID-19 and healthy control subjects. Figure 1A (right panel) shows the data processing strategy. Dimensionality reduction analysis including all data sets, with complet hierarchical clustering, is shown in Supplementary Figure S1, where all data points that contributed to the PCA are shown grouped by disease severity and unit variance scaling was applied to rows and SVD with imputation is used to calculate principal components. In Figure S1, X and Y axis show principal component 1 and principal component 2 that explain 27.5% and 15.1% of the total variance, respectively (N = 88 data points). To heighten the differences between groups, in Fig. 1B we present the principal components analysis (PCA) and hierarchical clustering of averaged data points. The X and Y axes show principal component 1 and principal component 2 which explain 67.6% and 21.6% of the total variance, respectively. Data was processed by ClustVis software for both Figire S1 and Fig. 1B.
Overall, the data indicated that Long-COVID is a distinct disease compared to COVID-19 ICU patients (severe disease), although the cohorts did share some mechanisms. In contrast, the COVID-19 medical ward inpatients (mild disease) shared mechanisms closer to the healthy control subjects.
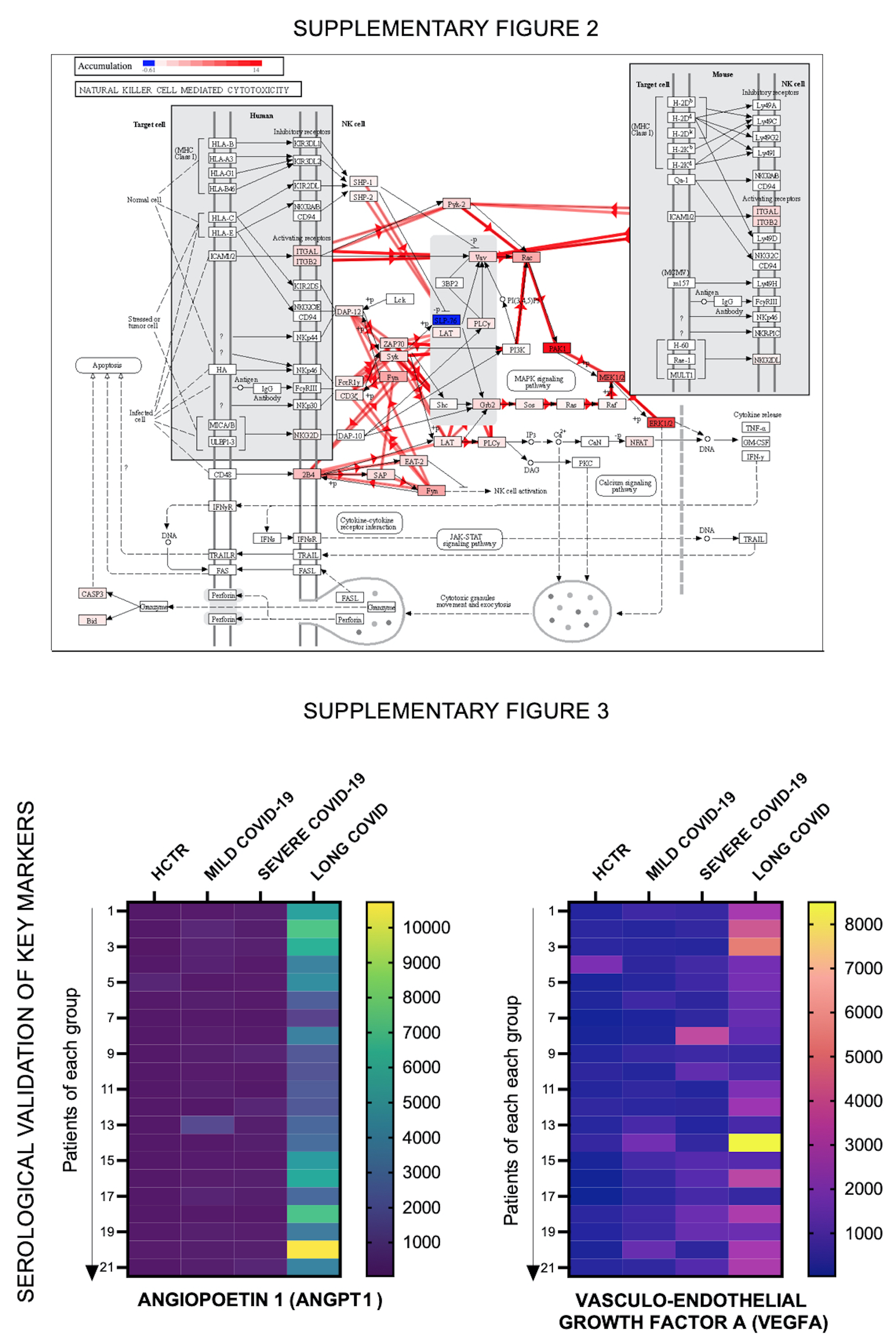
Natural killer (NK) cells from Long-COVID diseasechanged phenotye from activated to resting. The plasma proteome was deconvoluted to various immune cell-types from tissues based on their complex OMICS profile using the CIBERSORT analysis tool. The proportion of each cell-type in Long-COVID was compared to healthy control subjects (Fig. 2A). One major finding in Long-COVID outpatients is the shift in NK cells from activated to resting, indicating a shift in phenotype. In addition, memory B cells, memory CD4 activated cells and neutrophils were significantly elevated in Long-COVID, whereas resting dendritic cells, CD4 memory resting cells and M1 macrophages were down-regulated. The NK cell cytotoxic pathways, output of the iPathwaysGuide software, is shown in Fig. 2B. Overexpressed biomarkers are illustrated in red and the down-regulated ones in blue. The original NK-cell signaling KEGG map is presented in Supplementary Fig. 2. Investigations on the expression of individual markers showed down-regulations of SLP76, an immune response adaptor, while individual markers such as PAK1, MEK1/2 and ERK1/2 were upregulated and interconnected in signaling cascades that target the secretion of TNFα, IFNγ and GM-CSF. Furthermore, the VAV1 exchange factor for Rho-GTP-ases that connects directly to targets like ITGB2 and ITGAM was upregulated, thus predicting a subsequent increase in cellular adhesion and movement. Figure 2C (left panel) shows hierarchical clustering heatmaps, including NK cell signature markers (NCAM/CD56, killer receptors KLRK1 and KLRD1, IFNγ, IL-22, natural cytotoxicity receptor NCR1 and KIT stem cell factor). These above markers shared similarity in their behavior based on Pearson correlation algorithms (Fig. 2C, right panel), and all were upregulated in Long COVID (Fig. 2C, lower panel). Figure 2D (top left panel) illustrate the pathways analysis scoring with TNFα as the top hit; this graph shows the enrichment p-value on the horizontal axis (in a negative log scale) and the perturbation p-value on the vertical axis [48].
Prediction from the enrichment graph proved to be true, as the TNFα plasma expression was elevated in Long-COVID, as opposed to heathy control subjects (next plot, top row, Fig. 2D). The remaining plots in Fig. 2D portrayed elevated ANGPT1, whereas ANGPT2 did not change suggesting an ANGPT1/ANGPT2 conversion phenomen, as illustrated by the cartoon (Fig. 2D, bottom, right corner). The ANGPT1/2-Tie axis is critical regulator of endothelial inflammation and vascular leakage [49–51], and excess of ANGPT1 may reduce this process. Potential ‘vascular perturbation’ in Long-COVID pivoting around ANGPT/VEGF axis was validated by ANGPT1 and VEGFA measurement in plasma provided by a different group of patients than those approached by targeted proteomics (Supplementary Fig. 3, Heatmaps).
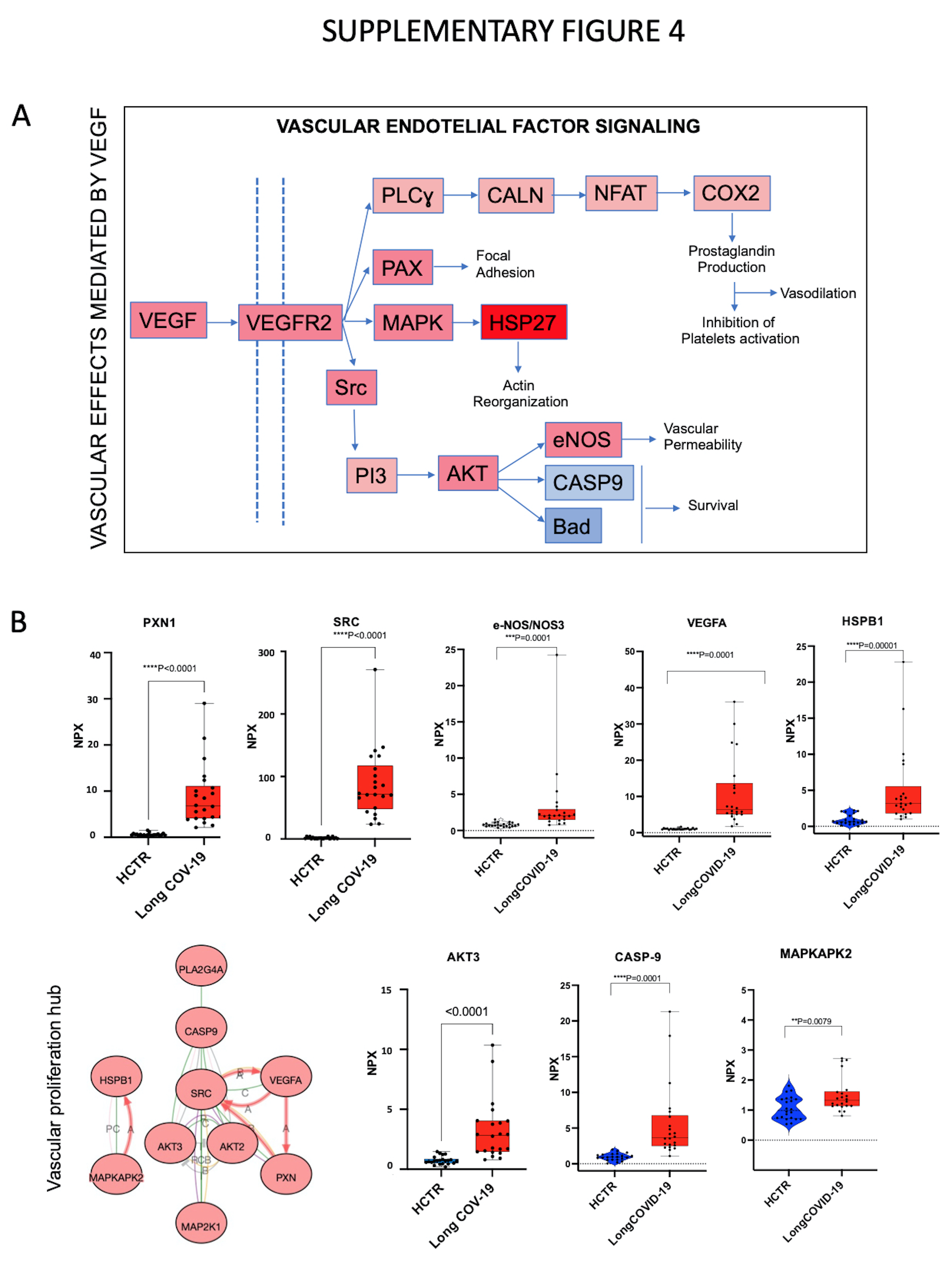
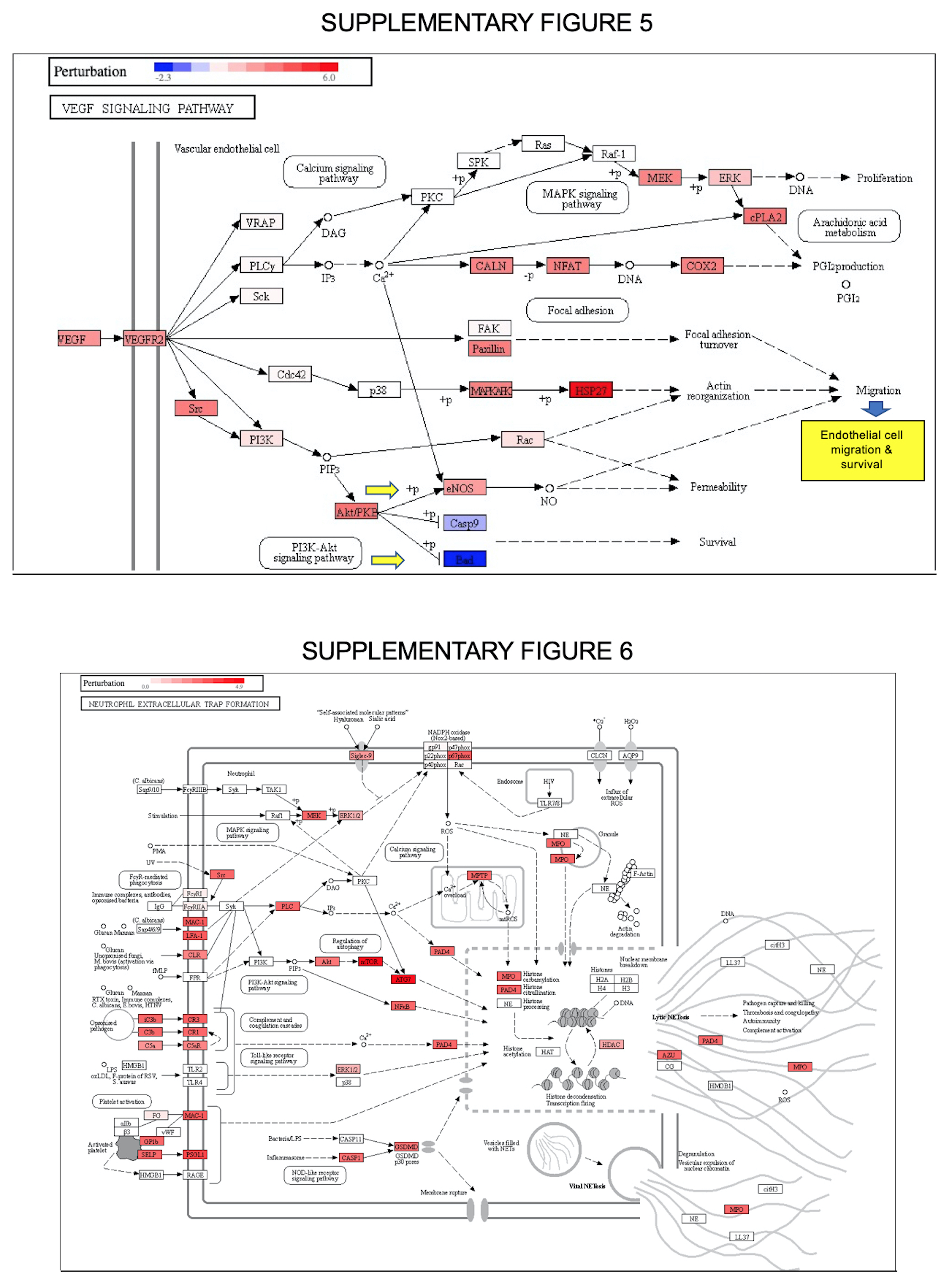
Resetting of immune-cell type proportions is reflected in vascular events mediated by VEGFA. Along with ANGPT1, VEGF signaling is also related to TNFα-linked pathways. Supplementary Fig. 4A shows that VEGFA pathway could regulate endothelial cell migration (signaling map is a crop-out of the KEGG chart presented in Supplemetary Fig. 5, as an output of iPathwaysGuide software). Supplementary Fig. 4C illustrates the upregulated markers that are members of the VEGFA pathway in Long-COVID, and included PXN, SRC, NOS3, and HSPB1. Other key markers such as Flt1 (VEGF receptor 1), AKT and MAPK, indicate endothelial cell survival, migration and proliferation. Key markers capable of protein-protein interactions are highlighted in the bottom left diagram and predicts VEGFA induced cell proliferation mediated by SRC and MAP-Kinases, and subsequent AKT signaling to influence cell survival and/or migration.
Neutrophil extracellular trap formation in Long-COVID. The features of the neutrophil extracellular traps (NETs) include: i) a defense mechanism against both micro-organisms and sterile triggers, ii) a DNA scaffold with granule-derived proteins, such as enzymatically active proteases and anti-microbial peptides, iii) an important role in inflammation, autoimmunity and other pathophysiological conditions (either detrimental or beneficial), and/or iv) it can be prompted by many triggers and via multiple distinct pathways with often unknown interrelationship [52–55]. The NET pathways shown in Fig. 3A represent a summary of the KEGG NET map output of iPathwayGuide, presented in Supplimentary Fig. 6. In red are the overexpressed biomarkers. Individual NET-specific/related biomarkers were featured in single expression graphs, where phospholipase (PLC1 and PLCγ), CAS1, and PDA4 were significantly upregulated in Long COVID, while NFkB, CR1, C3 and SLPG were down-regulated in this cohort when compared to severe COVID-19. While these results appear conflictual, our group and others previously reported a repurposed neutrophil phenotype in severe COVID-19 which may further differentiate in Long-COVID [52, 53].
Heatmaps representing the profiling of the neutrophil phenotype (based on a manually curated set of markers taken from specific literature reports) are presented in Fig. 3B [52–55]. The heatmaps show the hierarchical clustering of these markers in healthy control subjects and in COVID-19 patients along with their predicted interdependence (Pearson correlation algorhithm; Fig. 3B, right panel). Figure 3C highlights increased expression of neutrophil CD177, HLA-DR, ITGAM, ITGB2, and TLR2 manually curated markers in Long-COVID, as compared to the other patient cohorts. Interestingly, the expression of CD14, which is an innate immunity marker produced by macrophages and neutrophils was similar in both healthy control subjects and Long-COVID patients, but significantly down-regulated in mild and severe COVID-19 patients.
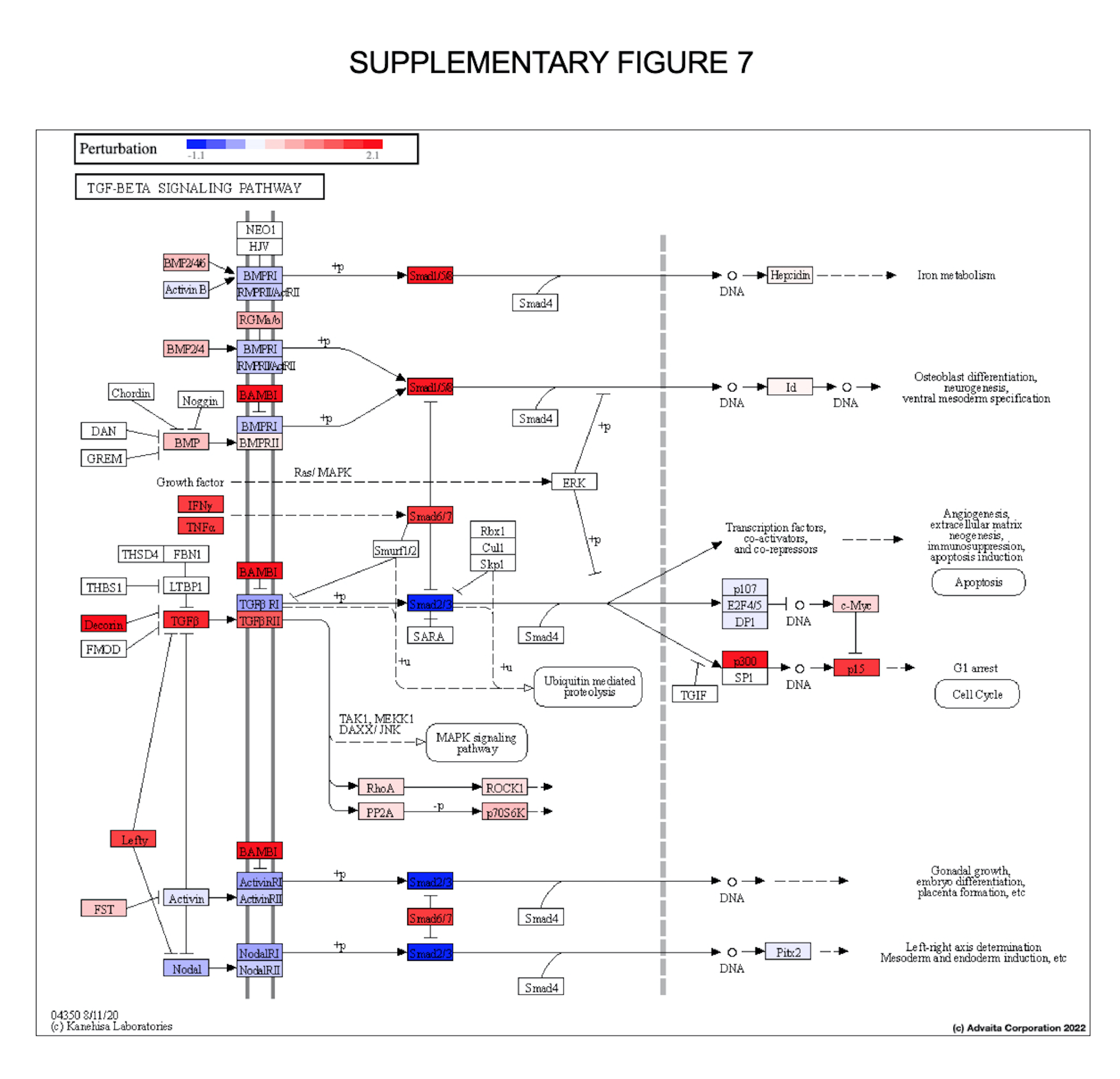
Silent TGFβ signaling and reduced EP300 favors vascular inflammation via TNF signaling and subsequent glucocorticoid resistance (GCR). Figure 4A illustrates TGFβ1 signaling pathway with both up-regulated (in red) and down-regulated (in blue) markers, per KEGG iPathwaysGuide output (Supplementary Fig. 7). Figure 4B graphs represent the expression of the individual markers associated with the TGFβ1 pathways. Key marker interactions are presented in the diagram (upper right). TGFβ1 and TGFBR2 were unchanged from healthy control subjects, thereby favoring TNFα pro-inflammatory signaling to induce an acute form of GCR [54, 56, 57]. TNFα has a significant and broad impact on the transcriptional performance of GR, but no impact on nuclear translocation, dimerization, or DNA binding capacity of GR [57]. A GR cofactor that interacts significantly less with the receptor under GCR conditions is EP/p300, a down-regulated biomarker in Long COVID that regulates the vascular bed via HIF under hypoxic conditions. EP/p300 strongly influences NFκB activation and thus, mediates inflamation [54]. EP/p300 knockdown reduces the transcriptional output of GCR, whereas its overexpression followed by NFκB inhibition reverts TNFα-induced GCR, trailing an authentic rearrangement model [57]. EP/p300 actions are quite vast, for instance it interacts with Smad1 to bridge coactivators such as NFκB transcribing IL-6, or it may interact with HIF1α/VEGFA axis [59, 60].
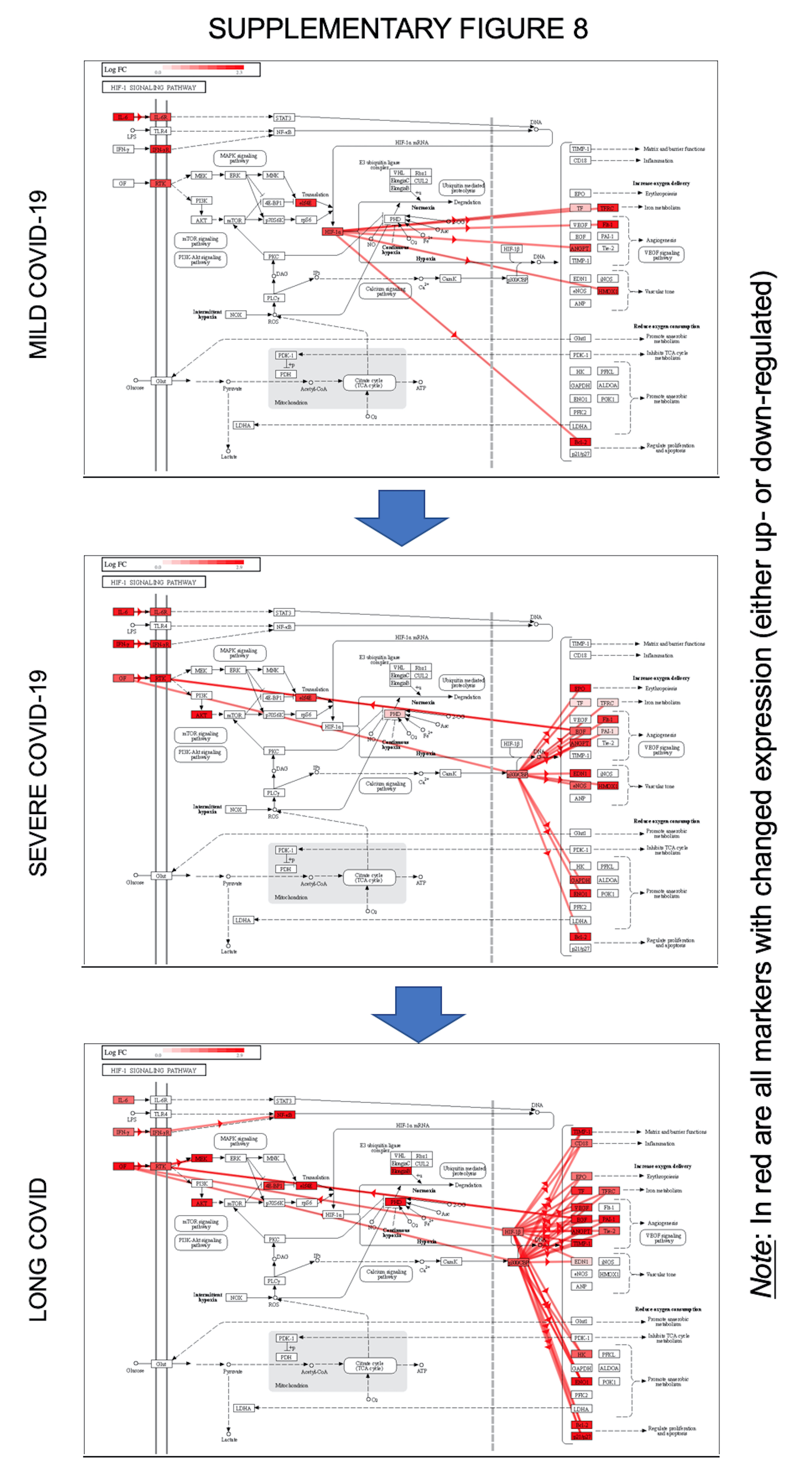
Vascular proliferation via HIF signaling pathway. The Long-COVID-19 proteome has been intersected with the mild and severe COVID-19 data sets, as well as the healthy control group, in a meta-analysis performed by iPathwaysGuide software. The resulted Venn diagram demonstrated that the Long-COVID group shared 80 signaling pathways among groups, 60 with the Severe COVID-19, but still retains 32 unique or independent pathways (Fig. 5A left diagram). Among all the pathways, HIF signaling was a major mechanism that progressed proportionally with disease severity, to a maximum in Long-COVID, as seen Fig. 5A and Supplementary Fig. 8 (where red arrows represent coherent cascades). HIF signaling in mild COVID-19 was initiated by IL-6 and STAT3, and targeted ANGPT and Flt1, followed by possible modifications in angiogenesis, iron metabolism (TFRC effector), vascular tone (HMOX1 effector), and cell survival (through BCL2 survival factor). In severe COVID-19, HIF signaling was triggered by IL-6, IFNγ and growth factors (VEGF and/or EGF) as ligands of receptor tyrosine kinases. Possible down stream modifications include erythropoietin (EPO), angiogenesis (Flt1, EGF, ANGPT1), vascular tone (eNOS, HMOX1), aerobic metabolism (GAPDH, ENO1) and survival (BCL2). HIF activation, and its consequences, in Long-COVID were significantly affected by the simultaneous decrease in (EP)p300, predicting extracellular matrix consequences via TIMP1, CD18-dependent inflammation (Integrin β2, ITGB2), and Tie2 regulated angiogenesis. HIF is a leading regulator of hypoxic/ischemic vascular responses, driving transcriptional activation of genes involved in vascular reactivity, angiogenesis, and the deployment of bone marrow-derived angiogenic cells. In parallel, (EP)p300 may function as a histone acetyltransferase with epigenetic functions reflected in endothelial cell proliferation and differentiation [54, 56–60], perhaps contributing to epigenetic modifications induced by SARS-CoV-2 infection. (EP)p300 may also affect the stimulation of hypoxia-inducible genes such as VEGF. While there are not therapeutic agents that target (EP)p300, it was predicted by the iPathwayGuide software that the HIF pathway can be however regulated by multiple re-purposed drugs presented in Fig. 5B.
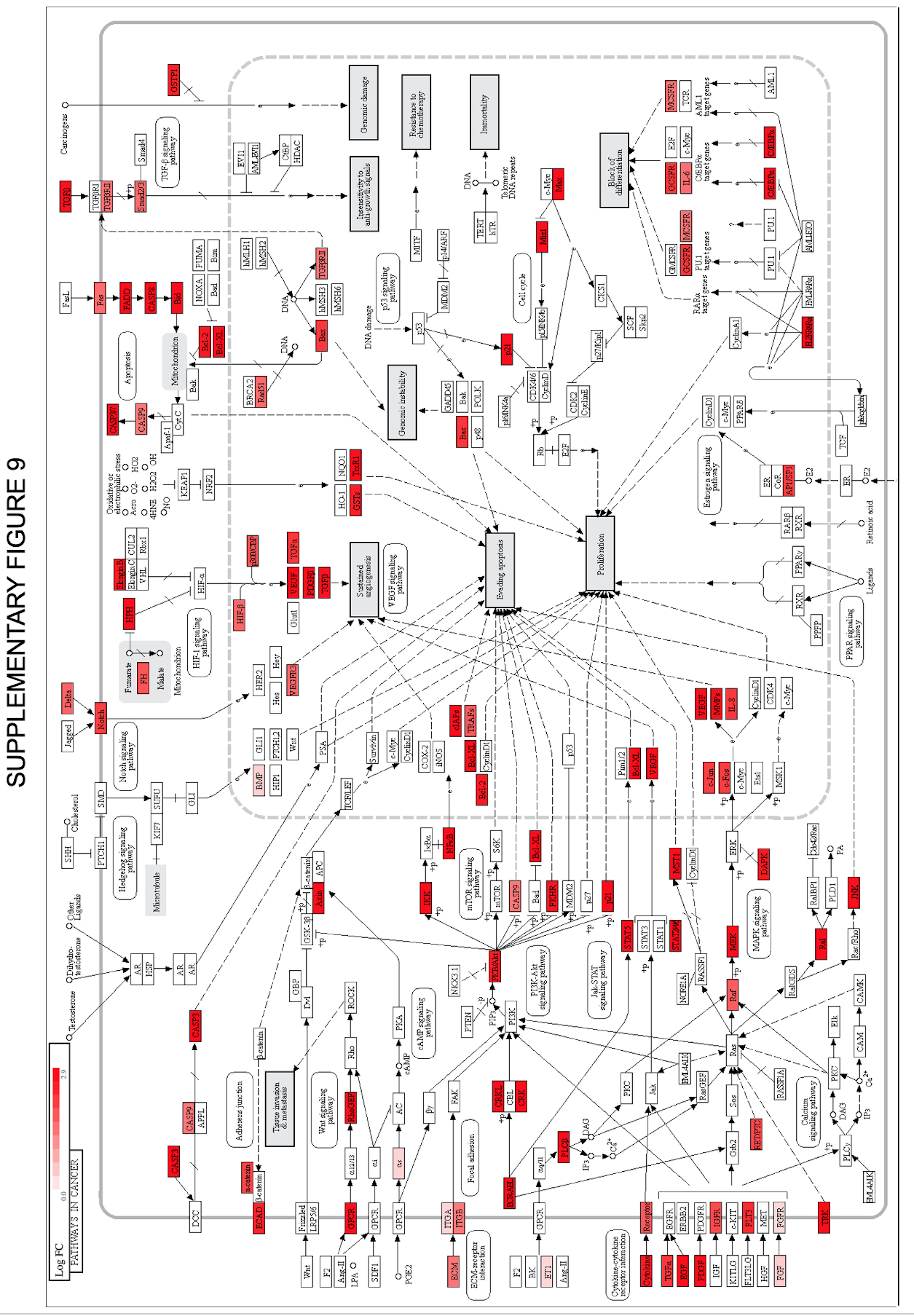
Proliferative pathways in Long-COVID pathology. To establish the nature of the vascular disease and its proliferative aspects, Long-COVID data was investigated with the KEGG cancer pathways (Supplementary Fig. 9), with the most representative signaling cascade led by HIF as highlighted in Fig. 6A (left panel). In this network, the up-regulated biomarkers are shown in red and down-regulated in blue. Plots in Fig. 6A (right panel) illustrate markers elevated in Long-COVID responsible for the sustainability of the vascular bed. Next, Fig. 6B heatmaps reflect the levels of growth-factors that support cell proliferation through RTK membrane receptors located within the vascular bed. Markers have been curated by OLINK. The diagram in the right panel illustrates correlations between these factors, in a similarity matrix based on Pearson correlation principles. Figure 6C graphs show the levels of the IGFBPs that regulate the bioavailability of IGF, a factor known to regulate both angiogenesis and sustainability of the vascular bed. Except IGFBP4, IGFBP6 and IGFBPL1, which seem Long-COVID specific, the other IGFBPs are common to severe COVID-19. The status of the IGFs and their binding proteins (IGFBPs) could reflect the tissue repair capacity.
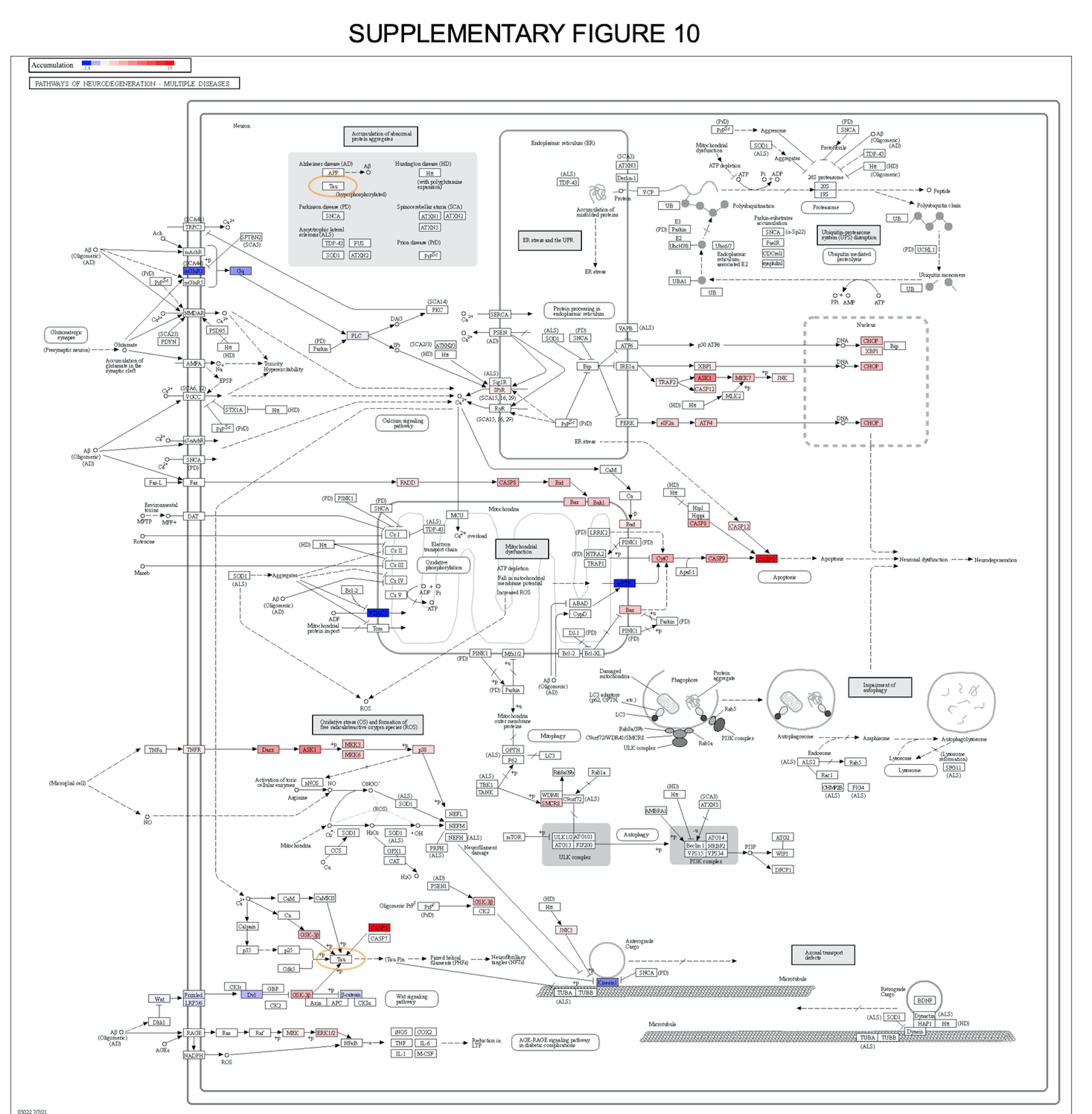
Long-COVID pathology implies brain dysfunction. The neurologic manifestations of the COVID-19 are well characterized and a comprehensive evaluation of the post-acute neurologic sequelae at 1 year was recently undertaken [61–64]. COVID-19 increased the risk of numerous neurologic sequelae such as ischemic and hemorrhagic stroke, cognition and memory disorders, peripheral nervous system disorders, episodic disorders (migraine and seizures), extrapyramidal and movement disorders, mental health disorders, musculoskeletal disorders, sensory disorders, Guillain–Barré syndrome, encephalitis, and/or encephalopathy. In Fig. 7A, hierarchical clustering heatmaps reflect the levels of neurological markers across the patient groups (markers have been curated by OLINK). The values of the PEA expression levels were hierarchically clustered based on Pearson correlation algorithms. Markers selected through the above methodology were investigated for functional annotation using tools from the GSEA platform and MSigDB data positories (Fig. 7B). This latest analysis demonstrated that functional clusters were formed around leukocyte migration, positive immune signals, glial cell differentiation, neurogenesis and MAPK regulatory modules. Taken together, these pathways predict a possible brain-blood barrier dysfunctionality grounded on cell proliferation.Graphs in Fig. 7C illustrate the expression levels of individual markers from the functional groups presented in Fig. 7B. One of the highly expressed markers, was the amyloid precursor protein (APP; Supplementary Fig. 10) which is known to be a pathognomonic marker for both Alzheimer disease and brain inflammation [61–65]. Additional markers for brain dysfunction include JAM2 (endothelial tight junctions protein), SNAPIN (a mediator of neuronal autophagy-lysosomal function in developing neurons), KCNH2 (potassium channel), S100A14 (involved in cell motility adhesion and growth), KIAA0319 (language impairment biomarker), and IROR1 (a receptor tyrosine kinase like orphan receptor 1, which regulates neurites growth in the central nervous system having also WNT-signaling pathway functions, and being crucial for the auditive apparatus maintenance).
Long-COVID disease implies cardiometabolic injury based on vasculo-proliferative events. We confirm and further clarify previousy discovered COVID-19 mechanisms excerted in the heart [66]. In Fig. 8A, hierarchical clustering heatmaps reflect the levels and the dynamics of cardio-metabolic markers across all patient cohorts. Markers were curated by OLINK and their expression levels were hierarchically clustered using Morpheus software tools. Topographically, on this map three critical clusters can be observed, and functionally annotated (Fig. 8B) by a standard screening using GSEA/MSigDB tools. This analysis also yielded three functional clusters. Taken in order from 1 to 3, clusters refer to i) extracellular matrix remodeling, ii) cell adhesion and motility and iii) angiogenesis (tube formation). These functional clusters were then investigated for protein-protein interaction (Fig. 8C), where it can be seen the that the extracellular matrix remodeling and cell-cell adhesion functions were dominated by integrins (ITGB1, ITGA5, ITGA1, ITGB6, ITGB1B1) which can potentially interact with fibronectin (FN1), filamin (FLNA) and calcium binding markers, essentially mediating Ca2+-independent - cell matrix interactions. Graphs in Fig. 8D represent individual marker expression levels in plasma. The integrin changes depicted in these graphs may result into disbalanced extracellular-matrix, leading to apoptosis of the endothelial cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}