Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analysis
Observation on the cell size of adaxial and abaxial petal epidermal cells with SEM revealed that cell size of adaxial petal epidermal cells increased gradually from S1 to S6 (Fig. 1a, Table S1), while that of abaxial petal epidermal cells enlarged gradually from S1 to S5, reached the peak at S5, and then greatly decreased from S5 to S6 (Fig. 1b, Table S1). What's more, based on TEM observation, it was found that vacuole occupied most area of petal cells in O. fragrans (Fig. 1c, Table S1), and vacuole size increased gradually from S1 to S6. At S2, vacuole size of cells was nearly two times as large as that at S1. With the cell development, vacuole size of cells at S6 was just one time larger than that at S2 (Fig. 1c, Table S1).
Bud appearance with treatments
Before treatment, the buds of all plants were at S1, and they were spindle-shaped, with unfurled outer bud scales and furled inner bud scales. Bud appearance remained the same under 23 °C treatment for 2 d, 4 d, and 6 d (H2, H4, H6), as well as the 19 °C treatment for 2 d and 4 d (L2 and L4). When treated with 19 °C treatment for 6 d (L6), the bud stage reached at S2. Buds of L6 became globular-shaped, the inner bud scales unfurled and the inflorescence was visible.
Illumina sequencing and de novo assembly of sequence reads
Reference transcriptome sequencing generated 164,753,984 raw reads and 110,307,366 clean reads (Table S2). A total of 152,247 transcripts with average length of 751 nt were obtained and 96,920 unigenes with average length of 873 nt were yielded after all clean reads were assembled (Table S3).
Functional annotation and classification
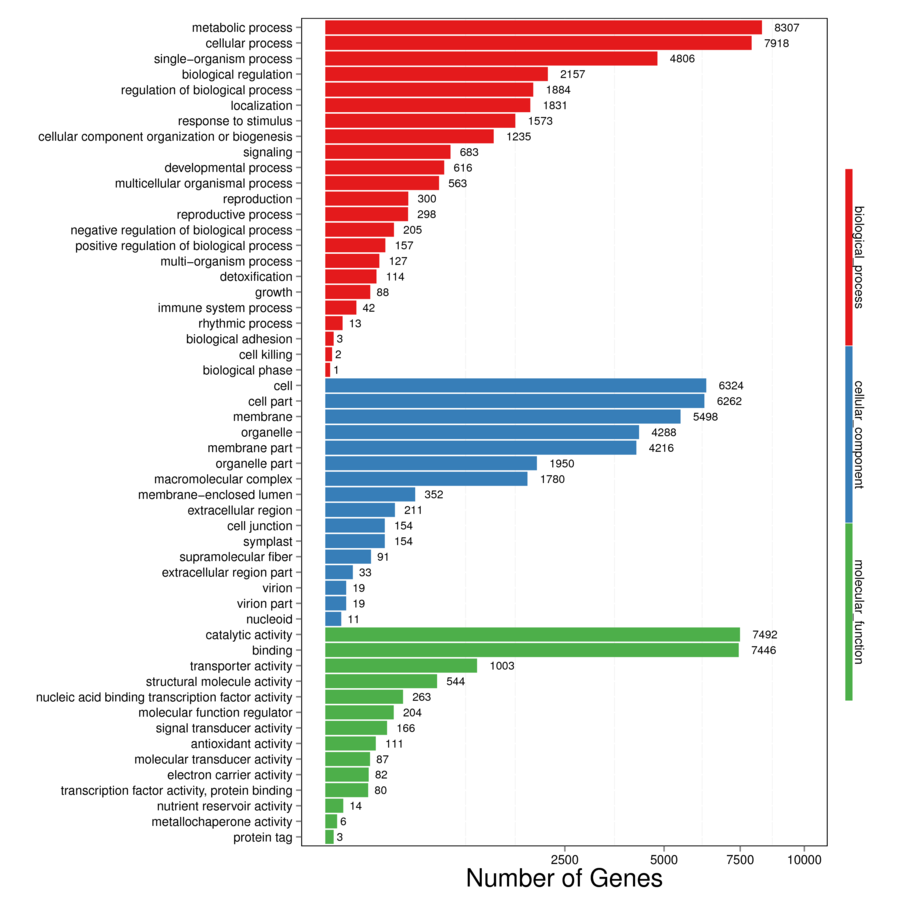
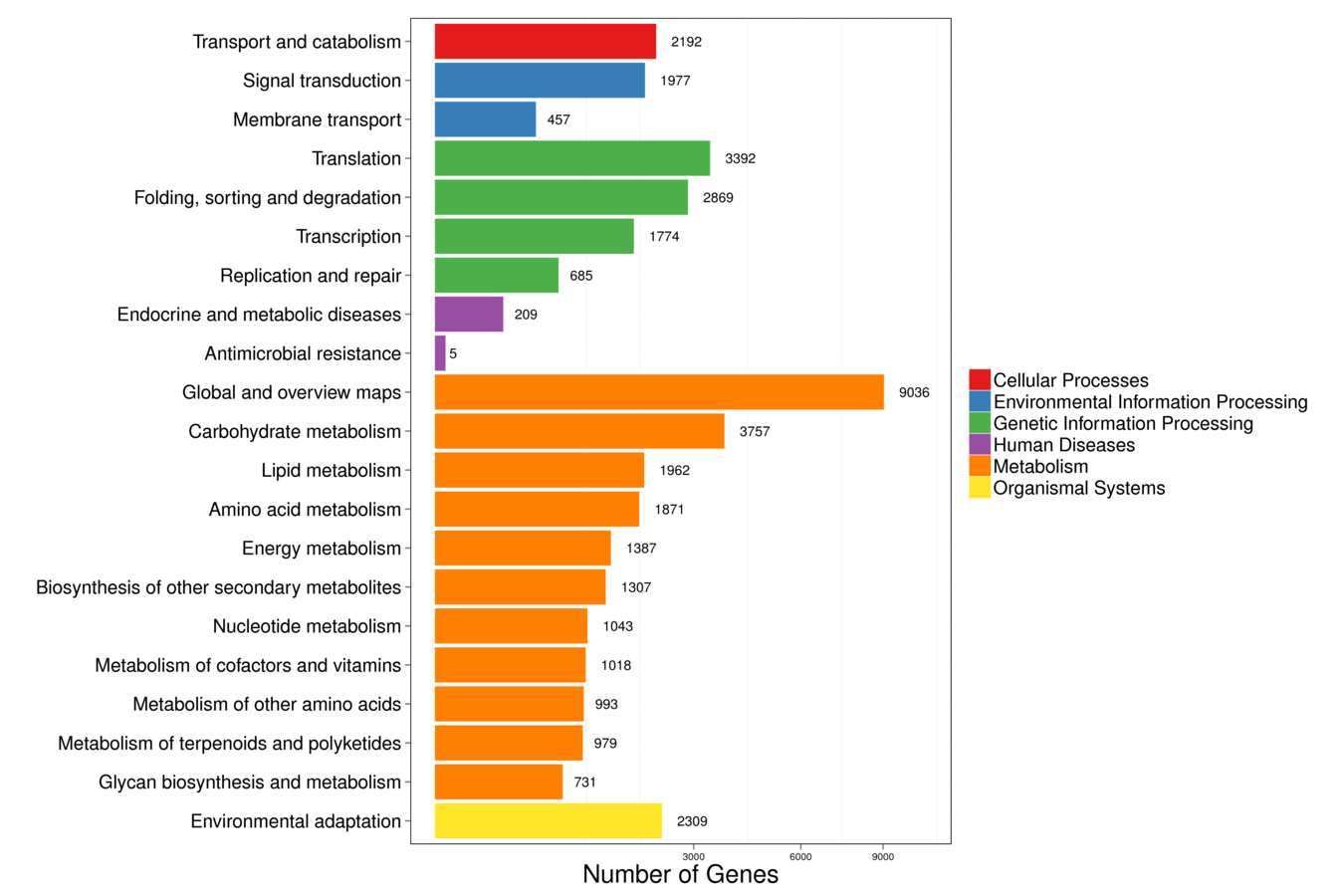
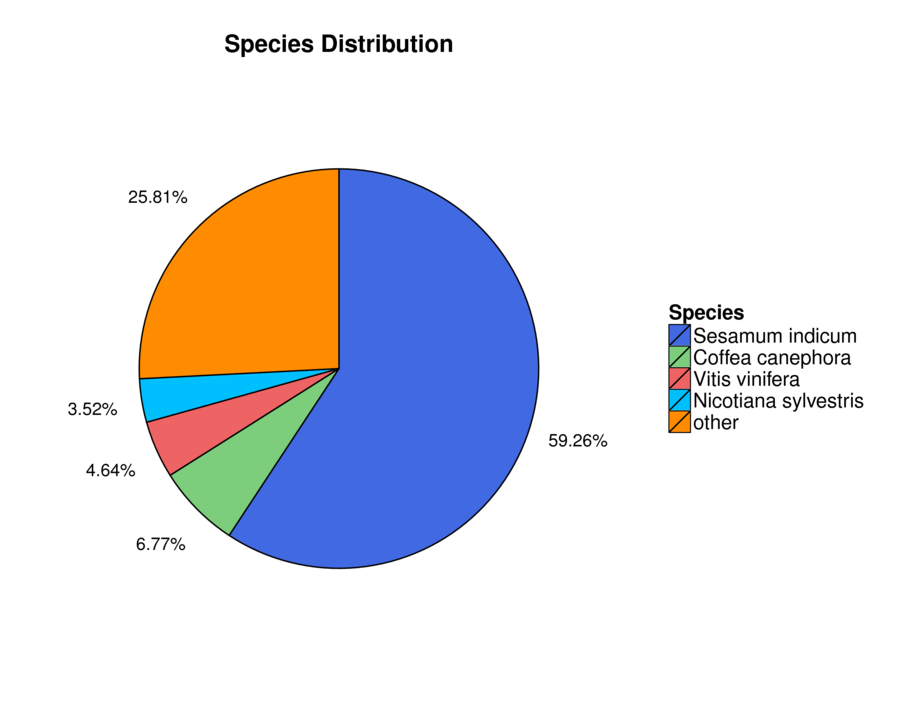
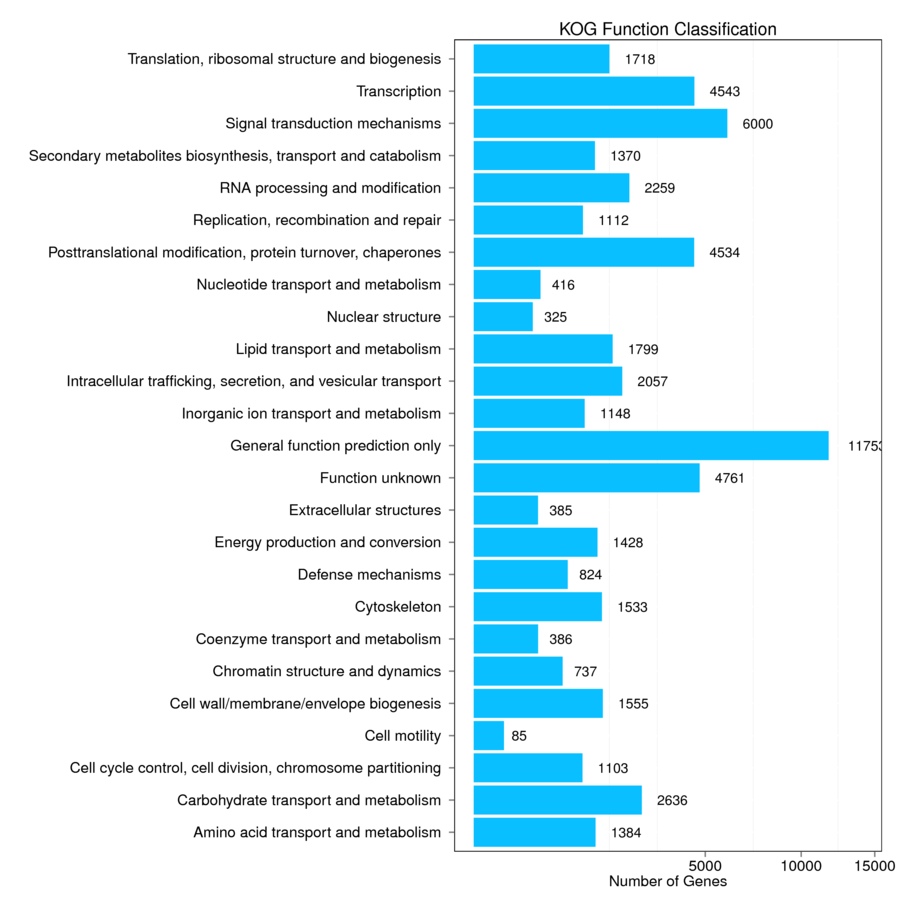
The assembled unigenes were aligned to seven databases (Nr, Nt, Swiss-Prot, KEGG, KOG, GO, and InterPro) in order to obtain the putative annotations (Table S4). Totally, 61,654 (63.61%) unigenes were successfully annotated using at least one database, while 8,150 (8.41%) unigenes were annotated using all seven databases. Total 57,721 (59.56%) of unigenes were annotated using NR database, 59.26% of which have high homology to sequences from Sesamum indicum (Fig. S1). In total, 37,284 unigenes were matched in the Swiss-Prot database, which is about 38.47% of all annotated unigenes (Table S4). There were 40,764 unigenes mapped into 134 KEGG pathways which can be divided into six large pathways including “cellular processes”, “environmental information processing”, “genetic information processing”, “human diseases”, “metabolism”, and “organismal systems” (Fig. S2). The pathways with the highest numbers of unigenes were “metabolic pathways” (Ko01100, 8,561 unigenes, 21%), followed by “biosynthesis of secondary metabolites” (Ko01110, 4,440 unigenes, 10.89%), “plant-pathogen interaction” (Ko04626, 1,876 unigenes, 4.6%), and “plant hormone signal transduction” (Ko04075, 1,444 unigenes, 3.54%) (Table S5). Next, GO analysis was performed and a total of 16,014 unigenes (16.52% of all annotated unigenes) were categorized into 53 GO terms under three main categories: biological process, cellular component, and molecular function (Fig. S3). Proteins related to “metabolic process”, “cellular process”, and “single-organism process” were enriched in biological processes. The “single-organism process” category defines a biological process involving only one organism, and the 4,806 unigenes annotated as such may be related to flowering organisms specifically. In the cellular component category, the “cell”, “cell part”, and “membrane” were the most highly presented GO terms. Under the molecular function category, the “catalytic activity”, and “binding proteins” were the most enriched (Fig. S3). We also performed KOG analysis to evaluate the functions of the assembled unigenes and 43,496 unigenes were assigned 25 KOG classification (Fig. S4). Among the 25 KOG classification, the cluster of “general function prediction only” (11,753 unigenes, 27.02%) represented the largest group, followed by “signal transduction mechanisms” (6,000 unigenes, 13.79%) and “function unknown” (4,761, 10.95%). “Cell motility” has the smallest proportion, only accounted for 0.20% (Fig. S4). Additionally, 46,405 (47.88%) unigenes in the transcriptome library were annotated against InterPro database (Table S4).
RNA-seq Illumina sequencing and mapping to the reference transcriptome database
Eighteen cDNA libraries for RNA-seq analysis were respectively sequenced, generating 23.58–24.14 Mb raw reads (Table S2). Furthermore, we obtained 23.58–24.13 Mb clean reads in 18 cDNA libraries, with the clean data rate of 99.94%-99.97%. Then, these clean reads were respectively mapped to the reference transcriptome database. Total mapped reads percentage and unique match percentage in the 18 libraries were similar. Total mapped reads percentage ranged from 86.39–88.44%, and unique match percentage ranged from 53.7–56.29% (Table S6). The correlation coefficients among biological replications are greater than 0.94 in all samples (Fig. 2, Table S7).
Screening and analysis of DEGs
We performed a detailed comparative analysis of the DEGs (P value ≤ 0.01 and |log2Ratio| ≥ 1) in three comparisons including L2 vs H2, L4 vs H4, and L6 vs H6. In the L2 vs H2 comparison, there were 7,365 and 15,864 unigenes were down-regulated and up-regulated, respectively. Meanwhile, 8,393 and 22,755 unigenes were down-regulated and up-regulated in the L4 vs H4 combination, respectively. In the L6 vs H6 comparison, 9,551 unigenes were down-regulated and 22,011 unigenes were up-regulated, respectively (Fig. 3a). In total, there were more up-regulated unigenes than down-regulated unigenes during floral opening under relatively low temperature. Additionally, 7,496 up-regulated DEGs were shared by three comparisons, while 4,108, 6,132, and 6,913 up-regulated DEGs were only up-regulated in the L2 vs H2, L4 vs H4, and L6 vs H6 comparison, respectively (Fig. 3b). 2,968 down-regulated DEGs were shared by three comparisons, while 2,954, 2,189, and 3,806 down-regulated unigenes were only down-expressed in the L2 vs H2, L4 vs H4, and L6 vs H6 comparison, respectively (Fig. 3c).
GO enrichment and KEGG pathway analysis of DEGs
GO analysis were used to classify the functions of the annotated DEGs under different temperatures treated for different days. In all comparisons, the most significantly enriched GO terms in biological process were “metabolic process”, “cellular process”, and “single-organism process”. In cellular component, five GO terms including “cell”, “cell part”, “membrane”, “membrane part”, and “organelle” were highly enriched. “Binding” and “catalytic activity” which were in the molecular function were significantly enriched in all comparison’s groups (Table 1). All DEGs were mapped to KEGG pathways in order to investigate the major pathways of them (Table 2). They were enriched in 132, 133, and 134 KEGG metabolic pathways in the L2 vs H2, L4 vs H4, and L6 vs H6 comparisons. In all the comparison group, “metabolic pathways”, “biosynthesis of secondary metabolites”, “plant-pathogen interaction”, “starch and sucrose metabolism”, and “plant hormone signal transduction” were the top five pathways with the largest number of DEGs (Table 2).
Table 1
The top ten most enriched GO terms of DEGs in all comparison groups
| GO term hierarchy 1 | GO term hierarchy 2 | Number of DEGs |
| L2 vs H2 | L4 vs H4 | L6 vs H6 |
| Biological process | metabolic process | 1823 | 2328 | 2539 |
| cellular process | 1693 | 2190 | 2331 |
| single-organism process | 1117 | 1424 | 1556 |
| Cellular component | cell | 1287 | 1626 | 1729 |
| cell part | 1271 | 1610 | 1710 |
| membrane | 1162 | 1395 | 1613 |
| membrane part | 886 | 1052 | 1229 |
| organelle | 866 | 1117 | 1187 |
| Molecular function | binding | 1628 | 2135 | 2266 |
| catalytic activity | 1693 | 2136 | 2379 |
Table 2
The top ten most enriched KEGG of DEGs in all comparison groups
| Pathway | ID | L2 vs H2 | L4 vs H4 | L6 vs H6 |
| Percent (%) | P-value | Percent (%) | P-value | Percent (%) | P-value |
| Metabolic pathways | ko01100 | 22.94 | 3.69895E-07 | 23.64 | 2.02306E-16 | 23.78 | 9.35908E-20 |
| Biosynthesis of secondary metabolites | ko01110 | 12.43 | 1.83429E-07 | 13.09 | 8.08594E-19 | 13.23 | 3.08193E-23 |
| Plant-pathogen interaction | ko04626 | 5.42 | 2.90029E-05 | 5.43 | 4.82036E-07 | 5.67 | 1.43759E-11 |
| Starch and sucrose metabolism | ko00500 | 4.03 | 9.30016E-08 | 3.69 | 3.81391E-05 | 3.77 | 8.46098E-07 |
| Plant hormone signal transduction | ko04075 | 3.96 | 0.01001351 | 4.27 | 6.50429E-07 | 4.65 | 2.91758E-15 |
| Protein processing in endoplasmic reticulum | ko04141 | 3.04 | 0.03015519 | 2.82 | 0.2699489 | 2.63 | 0.827178 |
| Carbon metabolism | ko01200 | 2.84 | 0.1309717 | 2.87 | 0.06273836 | 3.1 | 0.000191297 |
| Endocytosis | ko04144 | 2.72 | 0.5626473 | 2.93 | 0.07581657 | 2.73 | 0.5247731 |
| Ribosome | ko03010 | 2.76 | 0.006505746 | 2.94 | 4.06865E-06 | - | - |
| Phenylpropanoid biosynthesis | ko00940 | 2.55 | 2.74168E-13 | - | - | - | - |
| Amino sugar and nucleotide sugar metabolism | ko00520 | 2.51 | 0.007218067 | 2.66 | 1.27859E-05 | 2.46 | 0.003741373 |
| Biosynthesis of amino acids | ko01230 | 2.45 | 0.8067985 | 2.66 | 0.272135 | 2.78 | 0.04693815 |
| Spliceosome | ko03040 | - | - | 2.82 | 0.8755484 | 2.52 | 0.9998826 |
| RNA transport | ko03013 | - | - | - | - | 2.56 | 0.997384 |
Identification of DEGs involved in the cell wall metabolism
The expansion of petal cells depends on the cell wall loosening and cellulose biosynthesis, soluble carbohydrate allocation, ion and water transport, and cytoskeleton rearrangement [7]. Therefore, we explored the DEGs involved in the cell wall metabolism under different temperatures treated. EXP and XTH are involved in cell expansion, loosening and rearranging the cell wall fibers in growing tissues [8]. In our study, four α-EXP and one β-EXP were significantly different expressed under 23 °C and 19 °C comparisons (Fig. 4, Table S8). Among them, four α-EXP were up regulated under relatively low temperature (19 °C) and one β-EXP was down regulated under the treatment of 19 °C. Eleven unigenes which encoded as XTH protein were significantly expressed, seven of them (CL256.Contig3, CL3252.Contig2, CL10571.Contig1, Unigene1021, Unigene1631, Unigene24045, and Unigene25288) were up regulated under the treatment of 19 °C and four (CL1547.Contig1, CL7741.Contig1, CL8777.Contig1, and Unigene26777) of them were down-regulated under 19 °C treatment (Fig. 4, Table S8). Among them, the expression of CL8777.Contig1 decreased sharply in all three comparisons when the plants were exposed to relatively low temperature.
The expression of unigenes involved in cell wall synthesis, modification or hydrolysis which probably participate in the process of flower opening were also assessed (Fig. 4, Table S8). There were three cellulose synthase (CES) genes, six xylosidase (XYL) genes, two pectin esterase (PE) genes, five polygalacturonase (PG) genes, and two pectate lyase (PL) genes with dramatic change fold in all three comparisons (Fig. 4, Table S8). The expression of all CESs and PLs genes increased when treated with relatively low temperature of 19 °C for different days. Most of the XYLs were up-regulated except Unigene17352 under relatively low temperatures. Three PGs’ expression increased and two of them decreased when exposed to relatively low temperature. One PE gene was greatly up-regulated while one PE gene was down-regulated when the plants were exposed to 19 °C. More genes involved in cell wall synthesis, modification or hydrolysis are up-regulated, suggesting that these genes have vital roles in regulating petal cell expansion.
Aquaporins can facilitate the passage of water and/or small neutral solute fluxes across membranes and can be classified into four subclasses such as plasma membrane intrinsic proteins (PIPs), tonoplast intrinsic proteins (TIPs), nodulin-26-like intrinsic membrane proteins (NIPs) and small basic intrinsic proteins (SIPs) based on the sequence homology and cellular localization [24]. Three kinds of aquaporins including two PIPs, two TIPs and three NIPs, were found in our transcriptomes and most of them were significantly up-regulated under relatively low temperature (Fig. 4, Table S8). However, only the expression level of two NIPs (Unigene22675, and Unigene40148) decreased under relatively low temperature. Dehydrins are hydrophilic, thermostable stress proteins and some of them were identified as osmotic stress-responsive protein which could facilitate the inflow and outflow of water [25]. In our research, all the dehydrin genes were down-regulated when exposed to relatively low temperature (Fig. 4, Table S8).
Identification of DEGs involved in the phytohormone signal transduction pathway
There are some reports on phytohormones that can affect the floral opening [19], and therefore, we explored the DEGs involved in eight phytohormone signal transduction pathways. Nineteen unigenes were significantly differently-expressed under different temperatures in AUX signal transduction pathway which included the auxin influx carrier protein (AUX1), auxin/Indole-3-acetic acid (AUX/IAA), auxin response factor (ARF), auxin responsive GH3 family protein (GH3) and small auxin-up RNA family protein (SAUR) (Fig. 5). The majority of them had lower expression in relatively low temperature, especially three ARF members (CL661.Contig9, CL274.Contig1, and CL12908.Contig6), which decreased sharply as the treated days lengthened. Only one AUX1, two AUX/IAA, one ARF, and one GH3 were up regulated when treated with relatively low temperature (Fig. 5, Table S9). In JA signal transduction pathway, most of genes such as COI1, JAZ, and MYC2 were down-regulated, only two JAZ (Unigene43759 and CL8754.Contig3) genes and four MYC2 (CL37.Contig1, CL6240.Contig1, CL3114.Contig2, and CL2658.Contig2) genes were up-regulated under 19 °C treatment (Fig. 5, Table S9). Only six TGA genes in SA signal transduction pathway were expressed differently under different temperatures and two of them (CL2000.Contig1 and CL225.Contig1) were increased after 19 °C treatment (Fig. 5, Table S9).
Nineteen unigenes in the GA signaling pathway were also differentially expressed in different temperatures (Fig. 5). Five of six GID1 were down-regulated under relatively low temperature, with the exception of Unigene22649, which was nearly increased eight thousand times after 2 d treatment by 19 °C. Three DELLA and five PIF3 genes were decreased while two DELLA and three PIF3 genes were increased after relatively low temperature treatment (Fig. 5, Table S9). Only four of twenty unigenes including two PYR/PYL and two ABF (ABA responsive element binding factor) genes in ABA signal transduction pathway were up regulated after 19 °C treatment. However, the expression of one ABF (CL12556.Contig1) was decreased by two hundred times under the relatively low temperature at 19 °C compared with that under 23 °C (Fig. 5, Table S9). Most of the unigenes in CTK signal transduction pathway were down-regulated under relatively low temperature including all AHP, and all B-ARR members, especially the AHP (CL11134.Contig2), which was decreased severely after 6 d treatment by 19 °C (Fig. 5). However, most of the A-ARR members (CL7866.Contig2, Unigene18524, and CL8358.Contig1) were up-regulated when treated by relatively low temperature (Fig. 5, Table S9).
In ETH signal transduction pathway, the expression pattern of some ETR (Unigene14562), CTR1 (CL10648.Contig2), and EIN3 (CL10633.Contig1) genes, were more complicated. For example, the expression level of Unigene14562 and CL10633.Contig1 were decreased when treated by 2 d of relatively low temperature, while they were increased when the treatment time was lengthened. Most of the genes in this pathway were down-regulated after the relatively low temperature’ treatment (Fig. 5, Table S9). Eight of fifteen unigenes in the BR signal transduction pathways including all BRI1, one BSK, one TCH4, and one CYCD3 were increased under 19 °C treatment (Fig. 5, Table S9). The expression level of BRI1, especially Unigene37674 and Unigene35684 were dramatically increased when treated with 19 °C. However, seven of fifteen unigenes in this signal pathway were decreased under 19 °C treatment. The expression level of BZR1/2 were eight-fold under 19 °C compared to 23 °C, and the expression level were continuously increased as the treatment time prolonged. The same was true for TCH4 (CL8777.Contig2).
Identification of DEGs of transcription factors (TFs)
Many TFs changed dramatically under different temperatures, so we improved the screening criteria. The TFs with a |log2Ratio| ≥ 2 in any comparison were analyzed further. 79 TFs which were divided into eight gene family were selected for further analysis because they may be involved in petal cell expansion according to previous study (Fig. 6, Table S10). In the MYB TF family, only five out of twenty genes (Unigene36971, CL242.Contig2, CL10695.Contig2, Unigene11061, and Unigene30676) were up expressed under relatively low temperature. In the bHLH TF family, three genes (Unigene30441, CL796.Contig2, and CL6865.Contig1) were up expressed after treated by 19 °C. Among them, especially the bHLH79, the expression level of bHLH79 isoform X2 (CL6865.Contig1) increased five hundred times when treated by 6 d of relatively low temperature. However, the expression level of bHLH 117 decreased sharply when exposed to 19 °C. None of the TF genes was up-regulated under relatively low temperature in the AP2 and NAC TF family. In the TCP TF family, two genes (CL9936.Contig2, and Unigene10400) were up-expressed under relatively low temperature treatment. Four out of nine genes were up-regulated in the MADS TF family, specially the AGAMOUS gene, which is the C-function genes, and its expression increased significantly with the 19 °C treatment. In the WRKY TF family, only two genes (CL11608.Contig1, and Unigene860) were up-regulated under relatively low temperature. In Zinc finger TF family, four out of eleven genes’ expression increased when exposed to 19 °C. Generally speaking, only a small proportion of TFs were up-regulated under relatively low temperature.
qRT-PCR analysis
Ten differentially expressed unigenes comprising four genes involving in cell wall metabolism, four genes related to phytohormone signal transduction and two genes of TF were analyzed by qRT-PCR in order to confirm the results of RNA-SEq. All ten genes tested by qRT-PCR confirmed significant differential expression between 19 °C and 23 °C treatments (Fig. 7). Although fold changes of the ten genes between 19 °C and 23 °C treatments were not always similar in RNA-Seq and qRT-PCR results, the overall trend was consistent. Based on qRT-PCR results, the expression levels of CL10934.Contig2 (EXP), CL1473.Contig2 (XYL) and CL6542.Contig1 (PL) at 2d, 4d and 6d, were significantly lower in 23 °C treatment than those in 19 °C treatment, while the expression levels of other seven genes at 2d, 4d and 6d were higher in 23 °C treatment. Similarly, CL10934.Contig2, CL1473.Contig2 and CL6542.Contig1 were down-regulated, while other seven genes were up-regulated in 23 °C treatment, which verified the accuracy of RNA-Seq results.
{kind=link}
{kind=link}
{kind=link}
{kind=link}