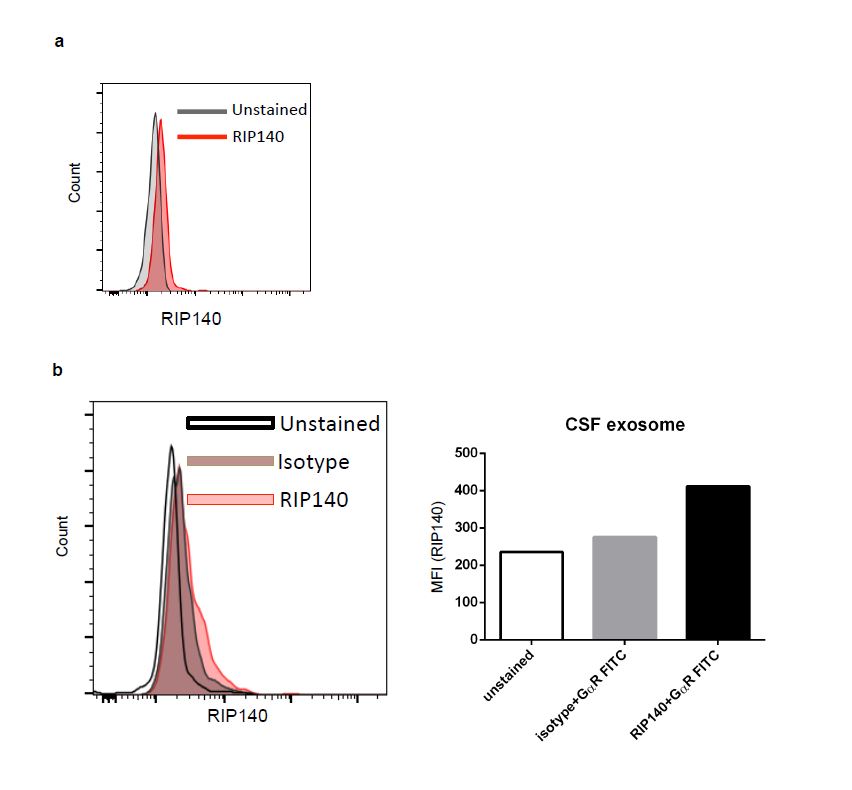
Our previous studies have established that RIP140, a nuclear transcription co-regulator, can be transported to the cytoplasm to augment specific cytosolic signaling pathways. It was very interesting that, in our preliminary experiments, we also detected RIP140 in mouse plasma (Fig. S1a). Careful studies comparing Crabp1 knockout (CKO) and wild type (WT) mice revealed that the plasma RIP140 level was higher in CKO mice as compared to wild type mice (Fig. 1a). In addition to plasma, RIP140-containing exosomes could also be isolated from cerebrospinal fluid (CSF). Importantly, CSF RIP140 levels were also higher in CKO than WT mice (Fig. 1b). These data suggested that Crabp1 could play a regulatory role in the secretion of RIP140 exosomes, which was further validated in molecular experiments (see later). To first demonstrate that the extracellular RIP140 was secreted from specific cells, we exploited a known RIP140-expressing cell culture system, the hippocampal neuron cell line HT22. This system allowed monitoring the secretion of RIP140. Clearly, RIP140 could be detected in the culture medium of HT22 (Fig. 1c). Herein RIP140 derived from exosomes was denoted as “exo-RIP140.”
We next determined whether the secretion of exo-RIP140 from neurons was a productive process, i.e. if exo-RIP140 can be taken by specific cells to exert biological activity, thereby mediating specific intercellular communication. We speculated microglia/macrophage as one type of cells receiving these neuronal exosomes, so we first determined whether exo-RIP140 of neurons could enter macrophages. In order to monitor exo-RIP140 derived from neuronal exosomes, we tagged RIP140 with GFP in donor cells (HT22) by expressing GFP-RIP140 in HT22 cells, and collected their conditioned medium (CM). The CM of HT22 was used to feed macrophage Raw 264.7 cells (Fig. 2a) and their cellular extracts were monitored on western blots. Fig. 2b showed that GFP-RIP140 was detected in the CM of the donor HT22 cells as predicted (upper panel). Importantly, the HT22 derived GFP-labeled exo-RIP140 was also detected inside the receiving macrophage (lower panel). This result unambiguously demonstrated that exo-RIP140 was secreted from donors (HT22 neurons) and entered the recipients (macrophages), showing transfer/mobilization of RIP140 between different cell types via exosomes.
To determine whether exo-RIP140, upon entering macrophage, remained biologically active, we exploited a well-established system where elevating RIP140 level in macrophages triggers their inflammatory polarization and dampens anti-inflammation. To conduct this experiment in a physiologically relevant context, i.e. monitoring exo-RIP140 specifically derived from endogenous RIP140 of donor neurons, we employed Crispr-cas9 to edit the endogenous RIP140 locus of HT22. Specifically we edited both alleles of endogenous RIP140 locus in HT22 by introducing GFP to tag endogenous RIP140 and adding early termination codons to delete (knock out) its coding region (Fig. 2c left), generating RIP140 KO HT22 cells. By performing transfer experiments similar to that described in Fig. 2a, we compared the effect of exo-RIP140 from wild type control (Ctrl) and RIP140 knockout (KO) HT22 (Fig. 2c right). We found that, for macrophage incubated with the CM of RIP140 KO HT22 (bars 3 & 4), their anti-inflammatory potential (induced by IL4, and marked by the expression level of Arg-1) (bar 4) was significantly elevated when compared to those cells incubated with the CM of ctrl HT22 (bar 2) (Fig. 2d, comparing bars 2 and 4), indicating that the IL4-induced anti-inflammatory status of receiving macrophages was elevated when the donor’s RIP140 (inflammatory) was knocked out. This result showed that exo-RIP140 remained biologically active upon entering macrophages. This experiment clearly showed that, neuronal exo-RIP140 could enter recipient cells (macrophages) and remain biologically active to affect their polarization. These results suggest a potentially global function of RIP140 in the whole animal by exosome-mediated transfer/mobilization to augment system-wide innate immune status. Since exo-RIP140 was significantly elevated in CKO mice (Fig. 1), we predicted that the CKO mice might exhibit systemic vulnerability to inflammatory stimuli (see following).
We systemically evaluated inflammation-related phenotypes of CKO mice as shown in Fig. 3a-c, including data reported earlier. These include increased high fat diet-induced obesity (Fig. 3a), and adipose expansion and hypertrophy (Fig. 3b), as well as insulin resistance [33], hyper-sensitivity to isoproterenol-induced cardiac hypertrophy that mimics inflammation [23], and altered anxiety and stress response (unpublished) (Fig. 3c). All these data support the notion of increased vulnerability of CKO mice in response to inflammatory stimuli on a system level. To examine whether this notion, that Crabp1 modulates the inflammatory status in animals, was clinically relevant, we performed clinical data mining of available patients data. It appeared that Crabp1 expression was dramatically and significantly reduced in multiple human inflammatory diseases including multiple sclerosis, lupus, inflammatory bowel diseases, vitiligo and psoriasis [34-38] (Fig. 3d). All these results together show that Crabp1 plays a role in regulating system-wide inflammatory response, at least partially, via modulating the mobilization of a pro-inflammatory regulator RIP140 through secreted exosomes.
To examine the underlying mechanism of the action of Crabp1 in regulating exosome secretion, we employed an in vitro system where both Crabp1 and RIP140 were endogenously expressed and CKO was readily available, an embryonic stem cell (ESC) line from CKO mice [16]. Since Crabp1 has a specific ligand, RA, we first examined if RA could affect the secretion of RIP140 from ESC. As shown in Fig. 4a, RA enhanced RIP140 secretion from WT ESC maintained in cultures without additional growth factors; importantly, this was abolished in CKO cells. Thus, RA acts on Crabp1 to regulate exosome secretion. To rule out potential effects involving gene transcription mediated by RARs, we used a transcription inhibitor, actinomycin D in the experiments. Fig. 4b showed that exo-RIP140 secretion was clearly independent of gene transcription. As a control, it involved protein expression (i.e., inhibition by cycloheximide). To further validate this RAR-independent activity of RA, we used a pan-RAR antagonist, AGN 193109, to block all RAR-mediated nuclear events in the experiment. As shown in Fig. 4c, AGN 193109 could not inhibit RA-induced exo-RIP140 secretion, further supporting a Crabp1-dependent activity of RA in regulating exo-RIP140 secretion. Indeed, an exosome secretion blocker (GW4869) dramatically inhibited the secretion of exo-RIP140, validating that RA/Crabp1-regulated RIP140 secretion was via exosomes (Fig. 4d).
Two signaling pathways could be directly targeted by Crabp1, the MAPK and CaMKII signaling pathways [19]. Interestingly, RIP140 exosome secretion was blocked by MAPK inhibitor U0126 (Fig. 4e), but not CaMKII inhibitor (not shown). Furthermore, two previously identified selective ligands of Crabp1 [15], C3 and C4, were also able to act on Crabp1 to regulate exo-RIP140 secretion as effectively as RA (Fig. 4f). These results validate that exo-RIP140 secretion indeed involves RA-Crabp1, and that this is mediated by a specific Crabp1 target, the MAPK pathway.
It is worthy of notion that, in the culture system where no additional growth factors were provided, RA/Crabp1 could activate MAPK signaling (Raf-Mek-Erk) via binding Crabp1 to block the initiating Raf kinase [14]. In a typical growth factor-activated MAPK signaling, it is most robustly activated by Ras. In this context, RA-Crabp1 acted to compete with Ras for Raf activation, thereby dampening Raf-Mek-Erk signaling in a normal physiological context [17]. Therefore, RA-Crabp1 is a negative regulator of growth factor-elicited MAPK signaling. CKO mice are deficient in this negative regulation of MAPK signaling, and their exosome secretion would be elevated as compared to WT. This is supported by the experimental data (Figs. 1-3).
We proposed a model (Fig. 4g) for the action of RA-Crabp1 in regulating exosome secretion, which contributes to system-wide modulation of physiological processes including inflammatory response. In general, growth factors or mitogen signals are the principal triggers of the MAPK signaling pathway [39] to stimulate exosome secretion [40-42], including RIP140-containing exosome. This would maintain a certain level of system-wide propagation of inflammatory potential. RA-Crabp1, via dampening the MAPK signaling, would provide a negative modulating mechanism in order to tone down global inflammation when needed. If Crabp1 is deleted or defected, as in CKO mice that lack this negative counter-balance mechanism, the vulnerability to inflammatory stimuli would be increased.
{kind=link}