Reagents and antibodies
Minimum Essential Medium (MEM) and High-glucose Dulbecco's modified Eagle's medium (DMEM) were purchased from Hyclone (Logan, UT, USA) for cell culture. Trypsin-EDTA Solution (0.25%) was purchased from Gibco (Grand Island, NY, USA). BCA Protein Assay Kit and RIPA Lysis Buffer were obtained from Beyotime Biotechnology (Shanghai, China). Pre-stained protein marker was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Jet PRIME Transfection Reagent was provided by Polyplus Transfection SA (Strasbourg, France). Vorinostat (purity > 99%) was purchased from Aladdin (Shanghai, China). N-acetyl-L-cysteine (NAC) was provided by Beyotime Institute of Biotechnology (Shanghai, China). Anti-POLR2J (16403-1-AP, 1:1000 dilution), anti-E-Cadherin (20874-1-AP, 1:1000 dilution), anti-N-Cadherin (22018-1-AP,1:1000 dilution), anti-EGFR (66455-1-Ig, 1:1000 dilution) and anti-Cyclin B1(28603-1-AP, 1:1000 dilution) antibodies were obtained from Proteintech (Wuhan, China). The anti-p-EIF2α (Ser51) (3398, 1:1000 dilution), anti-Cyclin A2(4656, 1:1000 dilution) and anti-Cleaved PARP (5625, 1:1000 dilution) antibodies were provided by Cell Signaling Technology (Danvers, MA, USA). The anti-STAT3(sc-482, 1:500 dilution), anti-p-AKT1/2/3 (Ser473) (sc-7985-R, 1:500 dilution) and anti-c-Myc (sc-40, 1:500 dilution) antibodies were provided by Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-p-STAT3(Tyr705) (ab76315, 1:1000 dilution) antibody was obtained from Abcam (Cambridge, MA, USA). The anti-GAPDH (db106, 1:10000 dilution) antibody was obtained from Diagbio. The anti-FLAG-tag (ABT2010, 1:10000 dilution) antibody was obtained from Abbkine, Inc. (San Diego, CA, USA).
Cell Culture
Glioma cell lines (T98G, U251, and A172) were obtained from Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). T98G was culture with MEM containing non-essential amino acids with 10% fetal bovine serum (FBS), U251 and A172 cells were maintained in DMEM with 10% FBS. All the cells were kept in a 37℃ condition with 5% CO2.
Quantitative reverse transcription-PCR (qRT-PCR)
The total RNA from cultured cell lines was extracted with Trizol reagent (Takara, Tokyo, Japan), and the concentration was measured with Nano-300 Nucleic Acid Analyzer (Allsheng, Hangzhou, China). Subsequently the total RNA was converted to cDNA by the HiScript II 1st Strand cDNA Synthesis Kit according to the manufacturers' recommendations (Vazyme Biotech, Nanjing, China). The qRT-PCR analysis was then conducted with HiScript II Q RT SuperMix (Vazyme Biotech, Nanjing, China). The 2 − ΔΔCt method was used to calculate the relative levels of target genes among groups. A list of all primer sequences is provided in supplementary Table 1.
Plasmid And Sirna Transfection
Cells were seeded into 6-well plates with a density of 1.0 × 105 cells per well. At 30–50% confluence, siRNA was transfected using jetPRIME transfection reagent (Polyplus Transfection SA, USA). siRNAs were obtained from GenePharma (Shanghai, China) and the sense sequences were shown as follows: siPOLR2J-1, 5’-AGGACACCAAGGUACCCAAUGTT-3’, siPOLR2J-2, 5’-AGAAGAAGAUCACCAUUAACATT-3’, and negative control siRNA, 5'‑UUCUCCGAA CGUGUCACGUTT‑3'. The POLR2J plasmid was obtained from GenScript Biotech (Piscataway, NJ, USA).
Sulforhodamine B (SRB) assay
After 24 h siRNA transfection, U251 and A172 cells were cultured into 96-well plates at a density of 3 × 103 cells/well. At 30–50% confluence, cells were treated with 1–6 µM vorinostat at 37°C for 72 h. The following steps are performed as described previously [14]. The OD value was detected at 540 nm with a microplate reader (Bioteck, Winooski, VT, USA).
Colony Formation Assay
U251 and T98G cells (2 × 103/well) were seeded into 6-well plates for overnight and were then transfected with siRNA, the culture medium containing the siRNA was replaced every 2–3 days for 10–14 days at 37°C. Following the supernatant is discarded, and the 6-well plates were carefully rinsed with PBS at three times, and then fixed with 4% paraformaldehyde for 30 min and 1% crystal violet stained for 20 min at room temperature, then washed with PBS and photographed.
Cell Cycle Analysis
U251 cells were seeded into 6-well plates at a density of 12 × 104 cells/well. At 30% confluence, cells were transfected with siPOLR2J for 48 h at 37°C, and then cells were collected and fixed with cold 75% ethanol at -20°C for overnight and washed with 500 µl PBS at three times. Subsequently, 1ml U251 cell suspensions were hatched with 5 µl Propidium iodide (PI) solution for 5 min at room temperature and cell cycle analysis was then performed with a FACSCalibur flow cytometer (USA).
Cell Apoptosis Assay
U251 cells (2 × 104/well) were cultured in 6-well plates. At 30% confluence, cells were transfected with siPOLR2J for 24 h and treated with 2.5 mM NAC for 6 h, afterwards treated with 4 µM vorinostat at 37°C for 48 h. The cells were harvested, cleaned with cold PBS, and resuspended in binding buffer solution mixed with 5 µl annexin V and 5 µl PI and cultivated in the dark for 15 min according to the kit's (BD Biosciences, USA) instructions and the fluorescence property was analyzed with a FACSCalibur flow cytometer (USA). The Flowjo software was applied to measure the figures.
Wound Healing Assay
The transfected cells were plated in 24-well plates for overnight. Artificial wounds were created using 10 µl pipette tip, and then rinsed with 500 µl PBS for 2–3 times to remove cell debris. Cell culture medium without FBS were added and then wound distances were recorded by microscope as 0 h distance. After 24 h, wound distance was recorded, and the percentage of wound healing was measured as (the wound distance of 0 h − 24 h) / 0 h wound distance× 100%.
Transwell Assay
After 12 h of serum starvation, the transfected cells were collected and resuspended in the serum-free medium with the density of 3×105/ml. The Transwell insert membranes were coated with or without a Matrigel for invasion or migration assay, respectively. The upper chamber (8 µm pore size) was seed with 200 µl of cell suspension and a 600 µl medium with 20% FBS was added to the lower chamber. After 24 h, cells on the bottom surface of Transwell insert membranes were fixed with methanol for 15 min and stained with 1% crystal violet for 30min at room temperature. Images were photographed and measured by ImageJ software.
Western Blotting
Western Blotting
Cellular protein lysate buffering solution was applied for protein extraction. Treated cells were collected and dissolved with cold RIPA lysate and the protein samples were separated by using 8–15% SDS-PAGE and translocated onto a PVDF (Schleicher, USA) membrane, followed by blocked with 5% skimmed milk for 1 h at room temperature. The bands were incubated overnight with specific antibodies at 4°C. Afterwards, underwent 1 h incubation at room temperature with the corresponding secondary antibody. The bands were visualized using ECL detection system (Millipore, Germany).
Co-immunoprecipitation (Co-ip) Assay
U251 cells transfected with POLR2J overexpressed plasmid for 48 h were lysed with IP buffer and centrifugation at 120,000rpm for 30 min at 4°C. Afterwards, the cell lysates were divided equally hybridized with 20 µl mouse IgG magnetic beads and anti-Flag Immunomagnetic beads, respectively, for 16 h at 4°C with gentle shaking. Subsequently, standing on the magnetic stand for 5 min, add 500 µl of PBST (NaCl 136.89 mM, KCl 2.67 mM, Na2HPO4 8.1 mM, KH2PO4 1.76 mM, 0.5% Tween 20) to the above precipitate, and redispersed the beads by gently blowing, then flip the sample up and down for 5 min and removed the supernatant after magnetic separation and repeated the above steps 3 times. Finally, resuspend with SDS loading buffer and performed SDS-PAGE detection.
Statistical analysis
All data are presented as the mean ± SD. Statistical significance was determined by unpaired two-tailed Student’s t test or one-way ANOVA followed by Tukey’s post hoc test using GraphPad Prism 7 software. P-values < 0.05 were considered statistically significant.
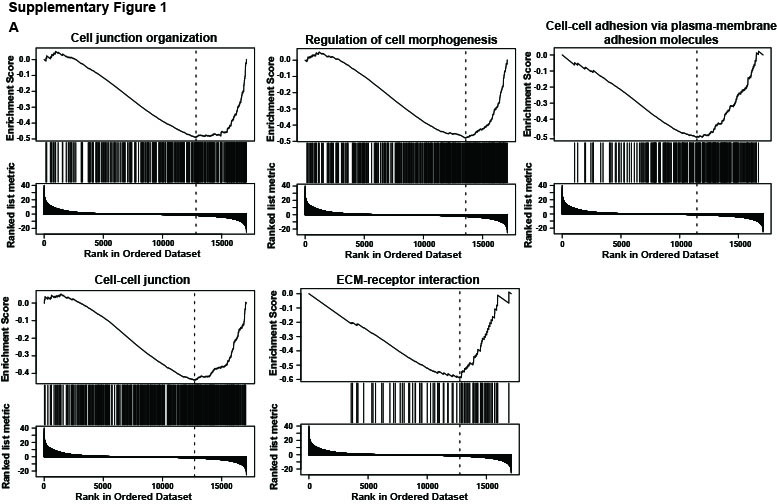{kind=link}