leukemia (CLL) is defined as a monoclonal proliferation at the expense of mature lymphocytes. It is the most common form of leukemia in adults in Western countries, accounting for 25% of all leukemias. 1
In the United States, 95% of CLL cases are of the B- cell phenotype, while in Asia, T-cell CLL predominates. 1.2
It is difficult to assess the true incidence, as it may be in patients with chronic lymphocytic leukemia Asymptomatic before diagnosis, asymptomatic was estimated to account for 30% of cases, but in the past 20 years the number of CLL cases presenting in asymptomatic stages has doubled from 30–60%, likely due to an increase in the number of blood tests being performed performed for other medical or surgical reasons, Moreover, sensitive techniques for diagnosing and differentiating CLL from other chronic lymphoproliferative disorders have only recently become routinely available. 3.4
The current estimated annual incidence in the USA ranges from 7,300 to 12,500 new cases, with an overall incidence of 2.3/100.00. 5.6
The Reasons
The cause is unknown, but much evidence points to a genetic component, such as the increased prevalence of CLL among first -degree relatives, the anticipation phenomenon, which shows an increase in severity, an earlier age with each generation, and an increased frequency of autoimmune disorders in relatives of patients with chronic lymphocytic leukemia. 7.9
No relation was shown to environmental factors, such as ionizing radiation, chemicals (gasoline and solvents from the rubber industry) and drugs. 10.11
Molecular Biolog
Chronic lymphocytic leukemia is a model of failed apoptosis BCL-2 family proteins are expressed ( which are key regulators of apoptosis ) are overexpressed in 90% of B-CLL cells, although in the vast majority of cases (96–99% ) there is no translocation involving BCL-2 .
Slow growing B-CLL cells accumulate in the body, mostly in the G0 phase of the cell cycle One consequence is acquired resistance to agents active in the cell cycle .
Imbalance in the ratio of key inducible and anti- apoptotic proteins, such as BAX and BAK (induction of apoptosis ), BCL − 2 (anti-apoptotic ), BAD, BIK, and HRK ( inhibitors of apoptosis ) It seems a They play an important role in the behavior and treatment response of CLL, although convincing clinical evidence is not yet available. 12.13
Mutations of the p53 tumor suppressor gene and increased levels of expression of the cyclin -dependent p27 kinase inhibitor have been shown to be associated with disease progression and overall poor prognosis. Impaired response to treatment with p53 mutants. 13.14
Cytokines are produced and released directly by CLL cells, such as tumor necrosis factor (TNF) and interleukin 8 (IL-8), as well as IL-2, which is produced by T lymphocytes and uptaken by CLL cells through specific receptors, involved in autocrine or paracrine loops and affecting CLL cell survival and proliferation. 15.16
IL-4 production is associated with increased expression of CD30. Since there is evidence that most CLL cells express CD30, this interaction may affect the environment of CLL cells and their immune functions. 17
One of the critical steps of the immune response to an antigen is the crosslinking expression of CD40 or CD154, which is produced by activated T cells, An upregulation of this ligand induced by CLL leukemic lymphocytes results in severe immunodeficiencies. 18
Cytogenetics :
There is no single identifiable cytogenetic abnormality in CLL, and the development of new techniques, such as fluorescent in situ hybridization (FISH), has increased the detection of chromosomal numerical and structural abnormalities. 1
The most common cytogenetic alteration is a deletion of 13q14 ( 51% ), followed by a deletion of 11q22q23
(17%-20%) Trisomy 12 (15% ), deletion 17p13. 19.22
there may be complex abnormalities; For example, CLL patients with trisomy 12 may have 13q 14 deletions. sync. 22.24
A number of cytogenetic abnormalities, including trisomy 12 and the presence of abnormalities on chromosomes 14q, 11q, and 17p, have been linked to poor treatment outcomes.
Trisomy 12 is associated with atypical lymphocyte morphology and immunophenotype (CD5–), FMC7 and disease progression .
Patients with 11q to be younger, with an advanced clinical stage at diagnosis With peripheral and abdominal lymphadenopathy mean, and a treatment-free period of nine months, as opposed to 43 months for those without deletion .
A chromosome 17 abnormality has been associated with a p53 mutation and fludarabine - resistant treatment and treatment failure in patients with Richter syndrome. and atypical CLL morphology, and it is unclear whether these cytogenetic abnormalities are primary or secondary events. 20.21.25.26
IgV gene mutations has been associated with decreased CD38 expression in a cohort of patients with good clinical outcome and improved survival. Therefore, CD38 may be useful as a surrogate for IgV genetic mutations and as a prognostic factor. 27.28
Clinical Manifestations :
About 40%-60% of patients with CLL are diagnosed in the absence of disease-related symptoms, even with very high circulating lymphocyte counts > 100 * 109/L.
Often, the presence of lymphadenopathy or an abnormal CBC during a routine medical exam is the only reason to suspect the diagnosis. 29
fever and may or may not be infected Have an infection or autoimmune disease. 30.31
Physical examination generally reveals painless, mobile lymphadenopathy or an enlarged spleen and/ or hepatomegaly. 1
Metabolic disorders (such as hyperuricemia) or mechanical disorders (such as airway obstruction) associated with tumor burden may also be present .
CLL cells can infiltrate any part of the body, including the skin and meninges; However, such findings are uncommon in chronic lymphocytic leukemia. 29
Myeloid insufficiency, particularly significant anemia ( hemoglobin < 11 g/dL) or thrombocytopenia (platelet count < 100 × 109/L ), was observed at diagnosis in 15% of CLL patients .
A positive direct antiglobulin test (DAT) was noted. It occurs in about 20% of patients at diagnosis but is not usually associated with hemolytic anemia. 32
Diagnosis:
The National Cancer Institute - Sponsored Working Group (NCI-WG) has published guidelines for diagnosis and response criteria for CLL. 33
-
Having at least 5 x 109 B lymphocytes/L in peripheral blood smear
-
An immunophenotype profile showing :
The aspirate should show bone marrow aspiration BM More than 30% of all nucleated cells are lymphoid. 33
Although BM examination is rarely required to make a diagnosis of CLL in general practice, it may be useful before treatment is initiated in order to identify prognostic factors .
BM examination is primarily indicated to assess response to treatment or to assess normals if unexplained anemia or thrombocytopenia is present. 33
Differential Diagnosis:
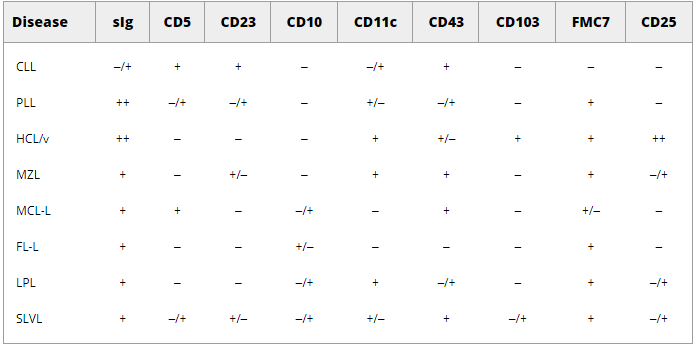
CLL can be distinguished from other closely related chronic lymphoproliferative disorders on the basis of morphology and immunophenotype (Table 1), such as (MCL-L) and follicular lymphoma (FL-L), PLL, hairy cell leukemia (HCL), and splenic lymphoma with villous cell lymphoma (SLVL). 33.34
Table 1: Phenotype of B-cell chronic lymphocytic leukemia .
Flow cytometry is very useful in differentiating MCL - L from CLL The morphology of MCL is cleft small lymphocytes, and the immunophenotype is type B cell neoplasia
CD5+), CD19+, CD20+ ), but MCL can be distinguished from CLL based on their expression of cyclin D1 and lack of CD23 expression
In addition, MCL has more intense surface immunoglobulin J and CD20 expression than CLL .
Prognostic And Staging Factors:
Rai and Binet systems (Table 2 ) are the most common alarm and phase determination systems for CLL. 35.36
Lymphocytosis alone is not classified by the Binet system, and neither system includes splenomegaly alone .
Prognostic factors regardless of the clinical stage are:
-
Age over 55 years old .
-
male sex .
-
Black race .
-
clinical condition bad .
older and younger CLL patients in trait presentation, response rates, or response duration .
prognostic factors that may predict outcome include: 37
| survival (years) | Clinical manifestations | danger | stage | classification system |
| > 10 | Lymphocytes only (in blood and marrow) | low | 0 | Rai |
| 7 | Lymphocytosis + lymphadenopathy | Average | I |
| 7 | Lymphocytosis + splenomegaly and/or hepatomegaly ± lymphadenopathy | | II |
| 1.5 | Lymphocytosis + anemia ± lymphadenopathy ± splenomegaly ± hepatomegaly | high | III |
| | Lymphocytosis + thrombocytopenia (thrombocytopenia < / 100 x 109 liters) ± anemia ± lymphadenopathy ± splenomegaly ± hepatomegaly | | IV |
| > 10 | Less than 3 decades | | A | Binet |
| 5 | 3 decades or more | | B |
| 2 | Anemia and/or thrombocytopenia | | C |
Table (2): Rai and Binet systems .
Mixtures:
There are several typical complications of chronic lymphocytic leukemia, including infections, autoimmune leukopenia, and transformation into high-grade lymphoma. 40
Transformation of chronic lymphocytic leukemia into diffuse large B-cell lymphoma or Hodgkin 's lymphoma occurs in approximately 1% of patients with chronic lymphocytic leukemia annually, and thus may affect up to 16% of patients during the course of the disease. 41
is termed Richter's transformation and usually has a very poor prognosis when diffuse large B-cell lymphoma is associated with primary chronic lymphocytic leukemia. 42
NOTCH1 mutations in patients with chronic lymphocytic leukemia increases the risk of Richter's transformation. 43
B - cell lymphoma, such as rituximab plus CHOP (cyclophosphamide, vincristine, doxorubicin, prednisolone), or more treatment Offensives such as rituximab plus cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternated with methotrexate and cytarabine, etoposide, prednisolone, vincristine, cyclophosphamide, doxorubicin ( cytarabine, rituximab). 44.45
The duration of treatment response in Richter transformation is usually short, and allogeneic stem cell transplantation should be considered in all responding patients with an available donor. 46
allogenic transplantation usually fails to prevent disease progression. 46
Transformation of chronic lymphocytic leukemia into Hodgkin 's disease is a separate and rare entity that traditional chemotherapy against lymphoma achieves Hodgkin 's lymphoma is often long-term remission Hodgkin. 41
Patients with chronic lymphocytic leukemia are also at increased risk of developing secondary cancers. 47.48
This risk is higher in elderly patients and in patients with comorbidities and increases with each round of treatment. Immuno-chemotherapy increases the risk of developing secondary myelodysplastic syndromes or acute myeloblastic leukemia. 47
The incidence of secondary myelodysplastic syndromes and acute myelogenous leukemia after FCR treatment is 2–5% depending on age and exposure to growth factors. 49
Immunochemotherapy with bendamustine may be associated with a lower risk of secondary myeloid malignancies than fludarabine plus cyclophosphamide. 49
Patients with chronic lymphocytic leukemia are also at risk B immune hemolytic anemia (in 2–4% ), immune thrombocytopenic purpura (2–5% ), red blood cell aplasia (0 5 − 1% ) and autoimmune granulocytopenia (< 1%, Autoimmune leukopenia, particularly hemolytic anemia, can also be triggered by exposure to purine nucleoside analogues. 50
Treatment for autoimmune leukopenia depends on the type of autoimmune leukopenia and whether the patient also requires treatment for primary chronic lymphocytic leukemia. 50
Patients with chronic lymphocytic leukemia often develop septic complications .
Until recently, infection was among the most common causes of death in patients with chronic lymphocytic leukemia, and acute blood infections continue to occur when treated with newer drugs. 50
The type of infection depends on the type of anti-leukemia treatment: immunosuppressive therapies such as purine analogues or alemtuzumab, kinase inhibitors, and BCL-2 inhibitors may reactivate opportunistic infections such as Pneumocystis jirovecii, cytomegalovirus, or Herpesviridae ; 51 As for irrigation, toximab may lead to the activation of hepatitis B virus ; 52 Myelosuppressive chemotherapy or immunochemotherapy may increase the risk of bacterial infection. Patients on distinct anti-leukemia therapies should therefore receive appropriate prophylaxis such as co-trimoxazole, antiviral therapies, or growth factor support to prevent persistent neutropenia .
Treatment With Ivig :
Hypogammaglobulinemia is the most prevalent inherent immune abnormality in CLL, and immunoglobulin replacement therapy ( IgRT ) is an alternative for patients with hypoglycemia and recurrent bacterial infections. 53
Although administration of immunoglobulin either intravenously ( IVIg ) or subcutaneously ( SCIg ) significantly reduces the rate of bacterial infections for CLL patients, it has no effect on the incidence of non-bacterial infections or on the patient's overall survival. 54
Ig preparations currently used in CLL contain more than 95% IgG and as a result, IgA deficiency persists and IgM. Analysis of infection-related factors in CLL patients showed a stronger association between the primary infection and a common antibody deficiency, that is, low levels of IgG. and IgA or IgM, rather than isolated deficient IgG. 55
While IVIg formulations were originally developed for IgRT in antibody- deficient patients, higher doses have been found to be effective as anti-inflammatory therapy in patients with autoimmune or inflammatory conditions. 56
Various mechanisms of action responsible for the immune-modulating ability of high doses of IVIg have been identified, eg: direct and indirect inhibition of T-cell activation and allergy induction, impairment of BCR- and TLR - signaling on B cells, and inhibition of the mononuclear phagocytic system. 57
Interestingly, patients receiving IgRT that increases IgG levels above 9 g/L have been shown to show evidence of disease control, suggesting that higher doses of Ig may have anti-leukemic activity in CLL patients. 58
{kind=link}