Research Article
Obtaining of novel pyrazolo pyrimidine disazo dyes derived from 4-position and a theoretical and experimental comparison of their characteristic properties
https://doi.org/10.21203/rs.3.rs-2521954/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Ethyl acetoacetate
Pyrimidin
Spectroscopy
DFT
Electronic properties
The presence of the azo (–N = N–) group causes azo compounds to have an very important place in dyes and pigments. Although azo compounds are used in food [1], cosmetics [2], printing [3], pharmaceutical industries [4], colored plastics [5], polymers[6], advanced applications in biomedical studies and organic synthesis [7], the most common uses are is the textile industry [8].
Azo dyes are an important group of chromophores with various applications in science and industry. The color of azo dyes varies according to the aromatic groups attached to the azo group. In particular, the synthesis of azo dyes containing heterocyclic rings is of great interest [9–14]. The reason for this interest is the properties exhibited by heterocyclic azo dyes. Heterocyclic azo dyes have bright color, washing and sublimation fastness, and chromophoric power [15, 16]. It has also been found that heterocyclic azo dyes show antifungal, anti-convulsant, anti-inflammatory, anti-tubercular, DNA binding and analgesic properties [17–26].
Pyrimidines are important in heterocyclic chemistry in terms of their synthesis, reactions and biological activities. Pyrimidine ring is found in many pharmaceutical agents and natural products [27–30]. Pyrimidines are heterocyclic derivatives with broad spectrum activity. It has been shown in the literature that they have antibacterial, antifungal, anti-inflammatory, antitumor, antiviral, anti-tuberculosis, analgesic activity and act as an enzyme inhibitor, anti-Parkinsonian drug and antioxidant compounds [31–35]. Because of these properties, synthesis of pyrimidine-containing heterocyclic compounds and investigation of their properties have an important place in the field of chemistry.
In recent years, determination of the structural, spectroscopic and electronic properties of chemical compounds by quantum chemical computation methods has become very popular. In our previous experimental and theoretical studies, the spectroscopic, electronic and structural properties of some azo dye compounds have been reported [13, 36, 37].
In this study, we present in detail the results of experimental and theoretical studies for newly synthesized 3a, 3b, 3c, 3d and 3e (3a-3e) azo dye compounds. In this study, the molecular structure, UV-vis analysis, vibrational, and 1HNMR spectra of the newly synthesised 3a-3e azo dyes are calculated by using the quantum chemistry methods based on DFT/B3LYP level with the basis set of 6-31G(d).
GENERAL INFORMATIONS
Compounds used in the experiment were purchased from Aldrich and Merck. Because of their high purity, they were used in direct synthesis without purification. Solvents used in spectroscopic studies were also spectroscopic purity.
Infrared spectra were measured using the Perkin-Elmer UATR Spectrum Two (FT-IR) spectrophotometer. Proton nuclear magnetic resonance spectra were measured with the Bruker-Spectrospin Avance DPX 400 instrument in deuterated dimethylsulfoxide solvent in tetramethylsilane reference. The melting points of the dyes were determined with the Electrothermal 9100 capillary melting point device. Ultraviolet-visible absorption spectra were recorded in PG T80 + high performance dual beam spectrophotometer at 1x10-6 M concentrations in DMSO, DMF, accetonitrile, methanol, acetic acid and chloroform.
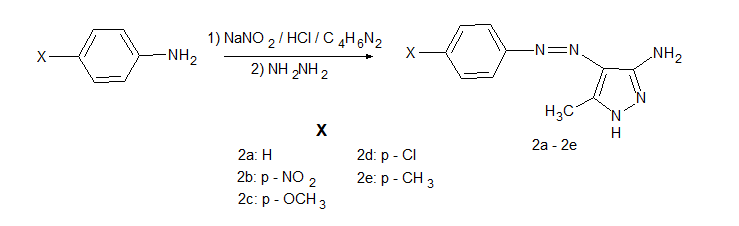
Synthesis of 5-amino-4-arylazo-3-methyl-1H-pyrazoles (2a-2e)
While 10 ml of HCl was added to 20 mmol of aniline and aniline derivatives and mixed magnetically in a salt-ice bath, a solution of 21 mmol of NaNO2 in water was added dropwise and left to stir for 2 hours. At the end of the period, the diazonium salts were obtained. In another beaker, 20 mmol of 3-aminocrotononitrile was dissolved in 15 ml of ethanol, 10 mL of water was added, and a solution of the coupling compound was prepared by adding 49 mmol of NaCH3COO. On the 3-aminocrotonitrile solution, the prepared diazonium salts were added dropwise and magnetically stirred in a salt-ice bath for 2 hours. The formed products were precipitated with water, filtered, dried and crystallized from a DMF-water mixture.
5,376 mmol 2-(arylhydrazone)-3-ketiminocrotononitrile compounds were dissolved in 30 ml ethyl alcohol in a 250 ml round bottom flask and heated with 30 mmol hydrazine monohydrate under reflux in a mantle heater for 4 hours. The compounds 5-amino-4-arylazo-3-methyl-1H-pyrazole (2a-2e) was synthesized. The synthesized dyes were precipitated with water again, filtered and dried. It was crystallized from a DMF-Water mixture.
Synthesis of 5-(4'-arylazo-3'-methyl-1H-pyrazol-5-ylazo)-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3a-3e)
Synthesis of 5-(4'-phenylazo-3'-methyl-1H-pyrazole-5-ylazo)-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3a)
7.46 mmol of the compound 5-amino-4-phenylazo-3-methyl-1H-pyrazole (2a) was dissolved in 15 ml of acetic acid and 15 ml of hydrochloric acid. While this solution was stirred in a salt-ice bath, a solution of 11.2 mmol NaNO2 in water was added slowly and stirred for 2 hours to form the diazonium salt. After this process, 20 ml of pyridine was added to 7.22 mmol of ethyl acetoacetate and dissolved, and the chelating component was prepared in this way. The diazonium salt obtained on the prepared coupling component was added dropwise and left in the ice bath on the magnetic stirrer for 2 hours to complete the coupling process. After 2 hours, the product formed was removed from the ice bath and precipitated with water at room temperature, filtered and dried. It was crystallized from a DMF-water (1:2) mixture.
The synthesized intermediate compound was dissolved in 60 ml of ethanol and heated in a mantle heater under reflux with 5.83 mmol of urea and 5.83 mmol of sodium ethoxide for 6 hours, then 5-(4'-phenylazo-3'-methyl-1H-pyrazole-5-ylazo)-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3a) was synthesized. The synthesized product was precipitated with water, filtered and dried. It was crystallized from a DMF-water (1:2) mixture. Yield: 64%, mp: 212–213 oC.
Synthesis of 5-[4'-(4''-nitro phenyl azo)-3'-methyl-1H-pyrazole-5-ylazo]-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3b)
Compound 5-[4'-(4''-nitro phenyl azo)-3'-methyl-1H-pyrazole-5-ylazo]-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3b) was synthesized 7.46 mmol of compound 5-amino-4-(4''-nitro phenyl azo)-3-methyl-1H-pyrazole (2b) using the method in section 2.3.1. Yield: 72% mp: 225–226 oC.
Synthesis of 5-[4'-(4''-methoxy phenyl azo)-3'-methyl-1H-pyrazole-5-ylazo]-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3c)
Compound 5-[4'-(4''-methoxy phenyl azo)-3'-methyl-1H-pyrazole-5-ylazo]-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3c) was synthesized 7.46 mmol of compound 5-amino-4-(4''-methoxy phenyl azo)-3-methyl-1H-pyrazole (2c) using the method in section 2.3.1. Yield: 67% mp: 229–230 oC.
Synthesis of 5-[4'-(4''-chloro phenyl azo)-3'-methyl-1H-pyrazole-5-ylazo]-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3d)
Compound 5-[4'-(4''-chloro phenyl azo)-3'-methyl-1H-pyrazole-5-ylazo]-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3d) was synthesized 7.46 mmol of compound 5-amino-4-(4''-chloro phenyl azo)-3-methyl-1H-pyrazole (2d) using the method in section 2.3.1. Yield: 72% mp: 233–234 oC.
Synthesis of 5-[4'-(4''-metyl phenyl azo)-3'-methyl-1H-pyrazole-5-ylazo]-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3e)
Compound 5-[4'-(4''-metyl phenyl azo)-3'-methyl-1H-pyrazole-5-ylazo]-4-hydroxy-6-methyl-1H-pyrimidin-2-one (3e) was synthesized 7.46 mmol of compound 5-amino-4-(4''-metyl phenyl azo)-3-methyl-1H-pyrazole (2e) using the method in section 2.3.1. Yield: 68% mp: 226–227 oC.
Computational Methods
In the theoretical part of this study, the quantum chemical computation-based GAUSSIAN 09 package program [38] was used to characterize the fundamental vibrational modes and structural properties of newly synthesized 3a-3e azo dyes molecules. Each of the theoretical work steps was carried out using the 6-31G(d) basis set at the DFT/B3LYP level.
In the first step, the optimization process was carried out to obtain the geometrical parameters such as bond lengths, bond angles and dihedral angles of the ground state stable structure of the synthesized 3a-3e azo dyes compounds with minimum energy. Based on these structural parameters obtained in the next step, the FT-IR spectrum of each molecule was calculated.
Theoretical chemical shifts calculation for 1H-NMR of the compounds is performed using by Gauge-Invariant Atomic Orbital (GIAO) method [39] in dimethylsulfoxide (DMSO) solvent. Solvent effect on calculated chemical shifts parameters are studied by means of the conductor-polarizable continuum model (CPCM) [40] provided by Gaussian 09 program. The 1H-NMR chemical shifts calculations are analyzed by taking Tetramethylsilane (TMS) as a reference.
Assignments of vibrational wave numbers for each molecule were made by potential energy distribution analysis (PED) using the Vibrational Energy Distribution Analysis (VEDA 4) [41] program. In order to compare the theoretically obtained harmonic frequencies for the synthesized disazo dye molecules with the experimentally observed frequencies, they were scaled with a correction factor of 0.960 [42].
The theoretical UV-vis spectrum analyzes for the synthesized azo dye molecules were performed separately for solvents with different polarities such as dimethylsulfoxide (DMSO), acetonitrile (CH3CN), acetic acid (CH3COOH), dimethylformamide (DMF), chloroform (CHCl3) and methanol (CH3OH).
On the other hand, boundary molecular orbital analysis was performed theoretically by using optimized structures of newly synthesized 3a-3e azo dye molecules, and HOMO-LUMO orbitals were visualized via GaussView 5.0.8 [43] molecular visualing program. Finally, the obtained results were compared with the experimental data.
Molecular Geometry
In this topic, molecular structures of synthesized 3a-3e azo dye molecules were prepared with Gaussian View 5.0.8 imaging program. Each of the molecules was optimized under the self-coordinated field principle approach. By determining the structural parameters such as bond lengths, bond angles and dihedral angles corresponding to the ground states of the molecules, the optimized geometrical structures of the molecules were determined. The theoretically calculated optimized structures of compounds 3a, 3b, 3c, 3d and 3e, respectively, are shown in Fig. 1. As a result of the calculation, it was determined that all the molecules have C1 group symmetry. The optimized molecular structures are non-planar.
Vibrational Analysis
The FT-IR spectroscopy plays an important role in the study and characterization of the spectra of molecules. Absorption bands in the IR spectrum guide us in determining the functional groups in the structures of the compounds. In this section, fundamental vibrational bands were determined for newly synthesized 3a-3eazo dyes using the DFT/B3LYP method and the 6-31G(d) basis set. Since the geometries of the compounds are non-planar, each has a 3N-1 vibrational mode.
|
Molecule |
ѵAr−H |
ѵN−H |
ѵAl−H |
ѵC=O |
ѵN=N |
ѵAr−H |
ѵN−H |
ѵAl−H |
ѵC=O |
ѵN=N |
|---|---|---|---|---|---|---|---|---|---|---|
|
Experimental |
Teoretical(DFT/B3LYP/6-31G(d)/ Scaled) | |||||||||
|
FT-IR Vibrational Frequencies Number of Wave (cm− 1). | ||||||||||
|
3a |
3061 |
3393 3280 |
2919 2850 |
1708 1668 |
1415 1369 |
3067 |
3503 3437 |
2941 2937 |
1754 1601 |
1420 1365 |
|
3b |
3063 |
3432 3314 |
2955 2921 |
1627 1605 |
1418 1380 |
3092 |
3499 3436 |
2941 2939 |
1757 1603 |
1419 1390 |
|
3c |
3075 |
3391 3193 |
2950 2922 2850 |
1707 1667 |
1429 1394 |
3077 |
3506 3438 |
3037 3027 3024 |
1753 1601 |
1422 1399 |
|
3d |
3047 |
3400 3293 |
2955 2921 |
1720 1622 |
1416 1380 |
3083 |
3502 3437 |
2991 2938 |
1755 1601 |
1402 1387 |
|
3e |
3073 |
3596 3395 |
2956 2920 2850 |
1713 1666 |
1415 1370 |
3082 |
3504 3437 |
2940 2937 2922 |
1754 1601 |
1405 1372 |
The fundamental vibrational modes of the C = O, N = N, NH, Al-H and Ar-H specific functional groups of the 3a-3e compounds are marked and compared with the experimentally obtained values and are presented in Table 1. As seen in Table 1, the frequency values obtained as a result of theoretical calculations are greater than the frequency values obtained as a result of experimental studies. This mismatch may be due to the phase studied, the neglected anharmonic effect, and the approximations used in the calculation. The theoretically obtained vibration frequencies were multiplied by scaling factor of 0.960 in order to eliminate the discrepancy between the theoretical and experimental results.
C-H Vibration Bands
It has been reported that, the vibration bands of the Ar-H groups in the aromatic rings show weak stretching vibration between 3100 to 3000 cm-1 in the IR spectrum [44, 45]. On the other hand, Al-H vibrational bands are observed as stretching vibrations in the region of the spectrum between 3000 − 2850 cm-1 [46]. In this study, the vibrational bands of the Ar-H group for 3a-3e compounds were calculated theoretically between 3067 and 3092 cm-1, and experimentally observed in the range of 3047 and 3075 cm-1. On the other hand, while Al-H was calculated between 2922 and 3037 cm-1 experimentally, it was observed between 2850 and 2956 cm-1 experimentally.
N-H Vibration Bands
The N-H stretching vibrations in the IR spectrum normally occur in the 3500 − 3200 cm-1 region of the spectrum. However, it was observed by Ganiyat et al. [47] in the region of 3250–3650 cm-1 in the IR spectrum. While the N-H stretching vibrations were experimentally observed in the range of 3436 and 3506 cm-1 in the IR spectrum recorded for 3a-3e compounds, they were marked in the range of 3193–3596 cm-1 in the calculation performed by the B3LYP/6-31G(d) method.
C = O Vibration Bands
Amost independently of other vibrations, the C = O bond is one of the most prominent and strong absorption bands. The main reason for this is the big change in the dipole. Due to the large dipole moment change of the carbonyl group between carbon and oxygen, all carbonyl groups exhibit a very intense and sharp peak in the 1800 − 1600 cm-1 region [48]. In our previous studies for azo dye compounds, we have seen both theoretically and experimentally that C = O vibration bands are in the range of 1700–1800 cm-1 [13]. For the A-E structures, the C = O (keto group) stretching vibrations were observed at the range of 1605–1720 cm-1 in the FTIR spectrum. On the other hand, the theoretically calculated C = O vibration bands A-E were calculated between 1601 and 1757 cm-1.
N = N Vibration Bands
N = N stretching vibration band is a kind of a very strong bond which is exhibit stretching vibration. In this study, the value of the N = N stretching vibrational mode is observed at the range of 1369–1429 cm-1 while the calculations recorded the range of 1365–1422 cm-1.
When correlations were made for the important vibrational frequencies found theoretically and experimentally for compounds 3a-3e, the correlation values were calculated as 0.9965 for compound 3a, 0.9960 for compound 3b, 0.9932 for compound 3c, 0.9982 for compound 3d, and 0.9954 for compound 3e. These correlation values indicate that the theoretical and experimental results are highly compatible.
NMR Analysis
Isotropic chemical shift values obtained in NMR spectrum analysis provide a powerful way to confirm or determine the molecular and magnetic properties of compounds. When the 1H-NMR spectra of the 3a-3e molecules were examined, three broad peaks were observed at 12.73–12.34 ppm (pyrazole NH), 12.96–12.77 ppm (pyrimidine NH), 12.06–11.77 ppm (OH). Other δ values were recorded as 2.34–2.30 ppm (pyrazole CH3), 2.56–2.54 ppm (pyrimidine CH3), and 8.27–7.03 ppm (aromatic H) as shown in Table 2. In addition, molecule 3c was showed δ values at 2.11 ppm (p-OCH3) and molecule 3e at 2.11 ppm (p-CH3).
We have seen in our previous studies that DFT- GIAO methods are very cheap and fast in terms of both cost and time in the theoretical calculation of 1H- NMR chemical shift values. The GIAO method is one of the most popular methods used to predict the isotropic chemical shifts of large molecules [49].
Using the optimized structures of the title compounds, the 1H NMR chemical shift values (with respect to TMS) calculations have been performed by B3LYP/6-31G (d) level by GIAO method. These calculations are conducted in DMSO-d6 solution. Theoretical and experimental chemical shifts values of 3a-3e compounds in 1H-NMR spectra is reported in Table 2. The R2correlation values between the experimental and theoretical chemical shift values of the compounds were examined. The correlation values were found as 0.9861, 0.9811, 0.9762, 0.9922 and 0.9922 for molecules 3a-3e, respectively. As it can be understood from the correlation value, the theoretical data are compatible with the experimental data.

s: singlet, m: multiplet, d: dublet, b: broad
Frontier Molecular Orbital Analysis
The HOMO-LUMO molecular orbitals and the energy difference (ΔE) between these two fundamental orbitals are very important as they play an important role in determining the activity of the molecule in chemical reactions. The HOMO orbital (Highest Occupied Molecular Orbital) represents the highest orbital occupied by electrons, while the LUMO orbital (Lowest unoccupied Molecular Orbital) represents the lowest unoccupied molecular orbital. The low energy difference between these two types of molecular orbitals means that intramolecular and intermolecular interactions will occur more easily [50].
|
Compounds |
3a |
3b |
3c |
3d |
3e |
|---|---|---|---|---|---|
|
EHOMO(eV) |
-5.91 eV |
-6.35 |
-5.62 |
-6.03 |
-5.82 |
|
ELUMO(eV) |
-2.91 |
-3.40 |
-2.79 |
-3.06 |
-2.86 |
|
ΔE (eV) |
3.00 |
2.94 |
2.84 |
2.97 |
2.96 |
|
I (eV) |
5.91 |
6.35 |
5.62 |
6.03 |
5.82 |
|
A (eV) |
2.91 |
3.40 |
2.79 |
3.06 |
2.86 |
|
χ (eV) |
4.41 |
4.88 |
4.20 |
4.54 |
4.34 |
|
η (eV) |
1.50 |
1.47 |
1.42 |
1.49 |
1.48 |
|
S (eV) |
0.33 |
0.34 |
0.35 |
0.34 |
0.34 |
As a result of the calculations performed for both molecules in this study, the energy difference between the HOMO-LUMO molecular orbitals of 3a-3e compounds was found to be 3.4344 eV, 2.9434 eV, 2.8427 eV, 2.972 eV and 2.9589 eV for 3a, 3b, 3c, 3d and 3e molecules, respectively. On the other hand, global descriptive parameters such as ionization potentials, electron affinities, chemical potentials, hardness and softness of molecules can be calculated based on the HOMO-LUMO orbital energies. The values of HOMO-LUMO orbitals of 3a-3e compounds and related electronic parameters are given in Table 3.
The regions on which the HOMO orbitals are localized give information about the nucleophilic character of the molecules, while the regions where the LUMO orbitals are localized give important information about the electrophilic character. The regions in which the HOMO-LUMO molecular orbitals are localized in the compounds are displayed in 3D with the help of the Gaussview molecular imaging program and are given in Fig. 2.
As can be clearly seen in Table 3, the lowest energy difference between the HOMO-LUMO orbitals belongs to the 3c compound with 2.84 eV. This means that the molecule with the highest chemical activity is the 3c molecule. It is clear from Table 3 that compound 3c is followed by compounds 3b with 2.94 eV, 3e with 2.96 eV, 3d with 2.97 eV and 3a with 3.43 eV, respectively.
UV-vis absorption studies (substituent and solvent effects)
In order to examine the dependence of the tautomeric balance of the molecules on the nature of the medium, the absorption behavior of the synthesized molecules in different solvents were investigated. For this purpose, the absorption spectra of the synthesized dye molecules were recorded in the range of 350–700 nm using DMSO, DMF, CH3CN CH3OH CH3COOH CHCl3 solvents (1x10-6 M). The results obtained were given in Table 4. 3d molecule was showed a single absorption peak in all solvents. It can be said that the 3d molecule exists in a single tautomeric form in all solvents. When Table 4was examined, it was seen that 3b, and 3e molecules gave an absorption peak and shoulder in DMSO when 3a molecule gave an absorption peak and two shoulders in DMSO. Moreover, it was observed that all molecules except to 3d molecule gave an absorption peak and shoulder in DMF solvent. From this point of view, it can be concluded that all synthesized molecules, except the 3d molecule, exist as a mixture of tautomeric forms in DMF and DMSO.
|
DMSO |
DMF |
CH3CN |
CH3OH |
CH3COOH |
CHCl3 |
Major Contribution* |
Minor Contribution* | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Dyes |
Exp |
Cal. |
Exp. |
Cal. |
Exp. |
Cal. |
Exp. |
Cal. |
Exp. |
Cal. |
Exp. |
Cal. |
CHCl3 | |
|
3a |
366, s 412 506,s |
400 |
366,s 412 |
400 |
364 398 |
399 |
398 366,s |
398 |
364 |
400 |
364 414 |
401 |
H->L + 1 (71%), H->L + 2 (14%) |
H-2->L (3%), H-1->L (7%), H-1->L + 2 (4%) |
|
3b |
366,s 464 |
477 |
366,s 462 |
477 |
434 |
476 |
420 |
476 |
368 |
476 |
400 |
487 |
H->L + 1 (71%), H->L + 2 (14%) |
H-2->L (3%), H-1->L (7%), H-1->L + 2 (4%) |
|
3c |
388,s 438 505 |
453 |
396,s 440 |
435 |
336 430 |
453 |
338,s 416 |
453 |
338 431 |
455 |
338 440 |
494 |
H-2->L (95%) |
- |
|
3d |
380 |
456 |
380 |
456 |
372 |
455 |
366 |
455 |
364 |
457 |
362 |
458 |
H-2->L (64%), H-1->L (30%) |
H->L (5%) |
|
3e |
370,s 432 |
472 |
370,s 432 |
473 |
370 398 |
472 |
370,s 399 |
449 |
372 |
473 |
370 424 |
457 |
H-2->L (95%) |
- |
|
* H:HOMO L:LUMO, s:shoulder | ||||||||||||||
When Table 4 was examined, it was observed that the absorption values of all molecules in DMSO and DMF solvents showed a bathochromic shift compared to chloroform. For example, the λmax value for the 3b molecule is 464 nm in DMSO, 462 nm in DMF, 400 nm in chloroform, and between 368 and 434 nm in the other solutions (Fig. 3a). This was an expected result since the dielectric constants of DMSO and DMF were higher than in other solvents. On the other hand, the theoretically calculated UV-vis absorption spectra of compound 3b in different solvents are given in Fig. 3b.
As can be seen from Table 4, the p-nitro derived dye (Molecule 3b) was shifted the absorption value into the bathochromic region in almost all solvents compared to 3a. A similar situation was also observed in the p-methoxy derivative dye(Molecule 3c). This result was thought to be due to the strong electron-withdrawing and electro-donating groups of 3b and 3c molecules.
The values of λ max for the samples in six different solvents were computed using the TD-SCF DFT/B3LYP/6-31G(d) method. Absorption spectrum calculations of the samples were carried out for the solvents of DMSO, DMF, acetonitrile, methanol, acetic acid and chloroform. The major and minor contributions from the HOMO, LUMO orbitals to transitions have been determined with the help of the GaussSum 3.0 software [51] by using the output file of the Gaussian 09W package software. The results obtained are compared with the experimentally observed results in Table 4. As seen in Table 4, the major contributions to electronic transitions at the absorption wavelength are HOMO -> LUMO + 1(71%), LUMO + 2 -> (14%), HOMO-1. -> LUMO (30%) and HOMO − 2 -> LUMO (95%). Table 4 also shows other wavelengths and the major and minor contributions corresponding to the electronic transitions in those wavelengths. The chloroform solution of the dye is chosen as reference while wave's contribution corresponding to electronic transitions in absorption spectra is calculated.
UV-vis absorption studies (acid and base effects)
While examining the acid-base effect on the absorption spectra of the molecules, the absorption spectra were obtained by adding 0.1 mol/L HCl and KOH to the solutions of the molecules in methanol. It has been observed that the absorption spectra of the molecules in methanol are extremely sensitive to base addition. When Table 5is examined, it is seen that the addition of KOH causes a bathochromic shift in λmax values for all molecules except the 3d molecule. For example, λmax for Molecule 3a was found to be 398 nm in methanol and 462 nm in alkaline methanol.
|
Molecules |
Methanol |
Methanol + HCl |
Methanol + KOH |
|---|---|---|---|
|
3a |
366 398 |
386 |
462 |
|
3b |
420 |
368 |
442 |
|
3c |
338 s 416 |
342 |
464 |
|
3d |
366 |
356 |
366 |
|
3e |
370 s 399 |
396 |
472 |
|
s:shoulder |
The λmax value of the 3d molecule remained the same in methanol solution and in alkaline methanol solution. The λmax values of the other 4 dyes showed a bathochromic shift in the basic medium. This result suggests that the tautomeric structures of all molecules except the 3d molecule in methanol and alkali methanol may be different. Figure 4 shows the acid-base effect on the absorption spectra of Molecule 3a.
When 0.1 mol/L HCl was added to the solutions of the molecules in methanol, it was observed that the λmax values of all molecules shift hypsochromic. For example, for Molecule 3a, λmax was 398 nm in methanol and 386 nm in asidic methanol.
We synthesized 5 new heterocyclic azo molecules based on 4-hydroxy-6-methyl-1H-pyrimidin-2-one. We characterized the synthesized molecules by elemental analysis and spectroscopic methods such as FT-IR and 1H-NMR. Again, we examined the absorption behavior of the synthesized molecules in different solvents and in acidic and basic environments.
On the other hand, theoretical calculations were performed for newly synthesized 3a-3e azo-dyes compounds using DFT/B3LYP method and 6-31G(d) base set in order to both support experimental studies and determine the electronic properties of the compounds. In this paper, it was determined that the correlations between the FT-IR data obtained theoretically and the FT-IR data obtained experimentally took values between 0.9982 and 0.9932. Similarly, it was evaluated that the correlation between the theoretical and experimental results of the 1H-NMR study took values between 0.9922 and 0.9762. These correlation values showed that the experimental results were in good agreement with the theoretical results. Therefore, it is quite clear that theoretical methods can be used as an important method for predicting experimental studies on azo dyes. On the other hand, from the HOMO-LUMO orbital analysis for compounds 3a-3e, it was determined that compound 3c had the lowest energy difference with 2.84 eV. This means that compound 3c has higher chemical reactivity than other compounds. The electronic absorption spectra (absorption wavelength λ(nm)) of the 3a-3e azo dye compounds were experimentally measured in six different solvents and calculated theoretically. The absorption wavelengths found and the major and minor contributions to the transitions calculated in the chloroform solution based on these are presented in Table 3.
Competing interests
- This manuscript is original.
- We are all aware of this manuscript’s content and approve its submission.
- The article has not been published anywhere.
- The article is not under consideration for publication elsewhere.
- No conflict of interest exists, or if such conflict exists, the exact nature of the conflict must be declared
- If accepted, the article will not be published elsewhere in the same form, in any language, without the written consent of the publisher.
Authors' contributions
Alpaslan Bayrakdar: Theoretical studies, Reviewing, Analysis interpretation, Data Curation and Writting. Aykut Demirçalı: Methodology, Experimental studies, Reviewing, Analysis interpretation, Data Curation and Writing. Hazal Kizak: Theoretical studies, Reviewing, Analysis interpretation and Data Curation. Pinar Tunay Tasli: Theoretical studies, Reviewing, Analysis interpretation, Data Curation and Writting. Fati Yıldırım: Methodology, Experimental studies, Reviewing, Analysis interpretation, Data Curation and Writing.
Declaration of Competing Interest: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding: The authors declare that there is no support for this study.
- J. P. Brown, G. W. Roehm, R. J. Brown, Mutat. Res. - Fundam. Mol. Mech. Mutagen. 56 (3), 249 (1978)
- H. Chen, Curr. Protein Pept. Sci. 7 (2), 101 (2006)
- H. S. Freeman, and J. Sokolowska, Rev. Prog. Color. Relat. Top. 29, 8 (1999)
- J. Rau, H.-J. Knackmuss, A. Stolz, Environ. Sci. Technol. 36 (7), 1497 (2002)
- R. M. Christie, Int. Polym. Process 34 (4), 351 (1994)
- B. J. Walekar, S. R. Mane, P. N. Bhosale, Adv. Polym. Technol. 36 (2), 243 (2017)
- H. Zollinger, Color chemistry: syntheses, properties, and applications of organic dyes and pigments. John Wiley & Sons: 2003
- A. A. Apan, and J. A. Peterson, Applied Geography 18 (2), 137 (1998)
- F. Karcı, and F. Karcı, J. Mol. Struct. 1024, 117 (2012)
- F. Karcı, and F. Karcı, Chem. Heterocycl. Compd. 49, 457 (2013)
- F. Karcı, F. Karcı, A. Demirçalı, M. Yamaç, J. Mol. Liq. 187, 302 (2013)
- T. Van Gog, and L. Kester, Cognitive Science 36 (8), 1532 (2012)
- F. Yıldırım, A. Demirçalı, F. Karcı, A. Bayrakdar, P. T. Taşlı, H. H. Kart, J. Mol. Liq. 223, 557 (2016)
- F. Yıldırım, A. Demirçalı, A. Ö. Kiraz, F. Karcı, J. Mol. Struct. 1222, 128850 (2020)
- Ç. K. Atay, M. Gökalp, S. Ö. Kart, T. Tilki, J. Mol. Struct. 1141, 237 (2017)
- G. Hallas, and J.-H. Choi, Dyes Pigm. 40 (2-3), 119 (1999)
- T. Tahir, M. Ashfaq, H. Asghar, M. I. Shahzad, R. Tabassum, A. Ashfaq, Mini-Rev. Med. Chem. 19 (9), 708 (2019)
- F.-C. Favre-Besse, O. Poirel, T. Bersot, E. Kim-Grellier, S. Daumas, S. El Mestikawy, F. C. Acher, N. Pietrancosta, Eur. J. Med. Chem. 78, 236 (2014)
- N. G. Khaligh, Dyes Pigm. 139, 556 (2017)
- A. M. Khedr, H. El-Ghamry, M. A. Kassem, F. A. Saad, N. El-Guesmi, Inorg. Chem. Commun. 108, 107496 (2019)
- B. Kirthan, M. Prabhakara, H. B. Naik, P. A. Nayak, E. I. Naik, Chem. Data Collect. 29, 100506 (2020)
- V. R. Mishra, C. W. Ghanavatkar, S. N. Mali, S. I. Qureshi, H. K. Chaudhari, N. Sekar, Comput. Biol. Chem. 78, 330 (2019)
- R. Raveendra, P. Prashanth, R. Hari Krishna, N. Bhagya, B. Nagabhushana, H. Raja Naika, K. Lingaraju, H. Nagabhushana, B. Daruka Prasad, J. Asian Ceram. Soc. 2 (4), 357 (2014)
- S. Sathiyavimal, S. Vasantharaj, T. Kaliannan, A. Pugazhendhi, Carbohydr. Polym. 241, 116243 (2020)
- J. D. Tadić, J. M. Lađarević, Ž. J. Vitnik, V. D. Vitnik, T. P. Stanojković, I. Z. Matić, D. Ž. Mijin, Dyes Pigm. 187, 109123 (2021)
- T. Tahir, M. Ashfaq, M. Saleem, M. Rafiq, M. I. Shahzad, K. Kotwica-Mojzych, M. Mojzych, Molecules 26 (16), 4872 (2021)
- I. Devesa, M. J. Alcaraz, R. Riguera, M. L. Ferrándiz, Eur. J. Pharmacol. 488 (1-3), 225 (2004)
- D. D. Dhavale, M. M. Matin, T. Sharma, S. G. Sabharwal, Bioorg. Med. Chem. 12 (15), 4039 (2004)
- C. Q. Huang, K. M. Wilcoxen, D. E. Grigoriadis, J. R. McCarthy, C. Chen, Bioorg. Med. Chem. Lett. 14 (15), 3943 (2004)
- P. Sharma, N. Rane, V. Gurram, Bioorg. Med. Chem. Lett. 14 (16), 4185 (2004)
- S. B. Bari, and N. G. Haswani, J. Saudi Chem. Soc. 21, S264 (2017)
- K. P. Cheremnykh, V. A. Savelyev, M. A. Pokrovskii, D. S. Baev, T. G. Tolstikova, A. G. Pokrovskii, E. E. Shults, Med. Chem. Res. 28, 545 (2019)
- B. Kumar, M. Kumar, A. R. Dwivedi, V. Kumar, ChemMedChem 13 (7), 705 (2018)
- N. Singh, S. K. Pandey, N. Anand, R. Dwivedi, S. Singh, S. K. Sinha, V. Chaturvedi, N. Jaiswal, A. K. Srivastava, P. Shah, Bioorg. Med. Chem. Lett. 21 (15), 4404 (2011)
- C. Türkeş, M. Arslan, Y. Demir, L. Cocaj, A. R. Nixha, Ş. Beydemir, Bioorg. Chem. 89, 103004 (2019)
- F. Karcı, A. Demirçalı, F. Karcı, I. Kara, F. Ucun, J. Mol. Struct. 935 (1-3), 19 (2009)
- P. T. Tasli, Ç. K. Atay, T. Demirturk, T. Tilki, J. Mol. Struct. 1201, 127098 (2020)
- A. Frisch, Wallingford, USA, 25p 470 (2009)
- K. Wolinski, J. F. Hinton, P. Pulay, J. Am. Chem. Soc. 112 (23), 8251 (1990)
- J. Tomasi, B. Mennucci, R. Cammi, Chem. Rev. 105 (8), 2999 (2005)
- M. H. Jamróz, Spectrochim. Acta, Part A 114, 220 (2013)
- M. Karakus, S. Solak, T. Hökelek, H. Dal, A. Bayrakdar, S. Ö. Kart, M. Karabacak, H. Kart, Spectrochim. Acta, Part A 122, 582 (2014)
- R. Dennington, T. Keith, J. Millam, Inc., Wallingford 20 (2008)
- M. Pagannone, B. Fornari, G. Mattei, Spectrochimica Acta Part A: Molecular Spectroscopy 43 (5), 621 (1987)
- M. Prasath, M. Govindammal, B. Sathya, J. Mol. Struct. 1146, 292 (2017)
- B. Smith, Infrared spectral interpretation: a systematic approach. CRC press: 2018
- G. K. Oloyede, P. A. Onocha, J. M. Oke, Am.-Eurasian J. Sci. Res. 6 (2), 116 (2011)
- G. Socrates, Infrared and Raman Characteristic Group Wave Numbers–Tables and Charts. John Wiley and sons, New York(2001)
- S. Sebastian, S. Sylvestre, N. Sundaraganesan, B. Karthikeyan, S. Silvan, Heliyon 8 (1), e08821 (2022)
- M. Ashfaq, K. S. Munawar, G. Bogdanov, A. Ali, M. N. Tahir, G. Ahmed, A. Ramalingam, M. M. Alam, M. Imran, S. Sambandam, J. Iran. Chem. Soc., 1 (2022)
- N. M. O'boyle, A. L. Tenderholt, K. M. Langner, J. Comput. Chem. 29 (5), 839 (2008)
Schemes are available in Supplementary Files section.
No competing interests reported.
- Scheme1.png
Scheme 1. Synthesis of 2a-2e compounds.
- Scheme2.png
Scheme 2. Synthesis of 3a-3e dyes.
{kind=link}
{kind=link}