5.1. The effect of temperature on extraction
Temperature is a complicated factor impacting solvent extraction processes. The dehydration of species enhances at high temperatures, while this factor reversely impacts the stability of metal complexes in an aqueous phase. Figure 1 illustrate the logD diagram to \(\frac{1000}{T}\) for both lanthanum and yttrium metals.
Figure 1. Graphs of logD against (\(\frac{1000}{T}\)) (K) for extracting of lanthanum and yttrium
As Fig. 1 depict, temperature rise decreases extraction, and this relationship is linear. The under-study system shows that the stability of the extracted D2EHPA ligand-rare earth elements complexes decreases as temperature enhances. The effect of this factor is more dominant at higher temperatures compared to the effect of the dehydration factor. Hence, the downward trend of extraction is achieved by temperature enhancement. Table 1 summarizes the thermodynamic parameters using the explained relations and results obtained from experimental studies. The extraction equilibrium constant displays that yttrium extraction by D2EHPA is more favorable than lanthanum extraction.
Table 1
Extraction thermodynamic parameters at 298 K
| Complex | Keq | ∆H (kcal/mol) | ∆Gexp (kcal/mol) | ∆S (cal/mol.K) |
| [La.NO3.2D2EHPA] | 6.46 × 10− 4 | -6.67 | 4.34 | -36.97 |
| [Y.(NO3)2.D2EHPA] | 467.68 × 10− 3 | -1.74 | 0.45 | -7.33 |
With regard to Table 1, ∆H values show the exothermic nature of lanthanum and yttrium extraction reactions. Accordingly, the enthalpy magnitudes of extraction reaction equal − 6,67 kcal/mol and − 1,74 kcal/mol, respectively, for lanthanum and yttrium complexes in the aqueous phase. Compared to yttrium, temperature variations have the highest effect on lanthanum extraction under the experimental conditions, such that as the temperature increases from 298K to 333K, the distribution coefficient of lanthanum decreases from 0.77 to 0.22. However, regarding the high extraction percentage of yttrium (> 99%), no sensible decline is observed due to temperature variations for this element, and extraction efficiency is still elevated at higher temperatures. The negative value of ∆S indicates that the degree of disorder decreases during the extraction of La(III) and Y(III) ions in a nitrate medium. This issue may be due to complex formation. The quantitative comparison between the entropy of lanthanum (-36.97 kcal/mol) and yttrium (-7.33 kcal/mol) extractions denotes that further disorder happens in the system due to yttrium extraction. This issue is one of the factors in the more selective extraction of yttrium than that of lanthanum. The positive values of Gibbs free energy confirm the non-spontaneous feasibility of the process and the unfavorable nature of the extraction reaction. As Table 1 displays, extraction by D2EHPA ligand is much more favorable for Y(III) (∆Gexp=0.45 kcal/mol) than La(III) (∆Gexp=4.34 kcal/mol).
5.2. Hydration Energy
The estimation of hydration free energy for ions is a challenging task since continuum dielectric solvent models are often insufficient when they deal with ionic solutes that have concentrated charge densities with strong local solute-solvent interactions. Researchers perceived that the published methodologies bring about systematic errors in the computed free energies because of the incorrect accounting of the standard state corrections for water molecules or water clusters present in the thermodynamic cycle [50]. With respect to the coordination of water molecules and nitrate anions in the first cluster surrounding every metal, we can consider the hydration reaction of yttrium and lanthanum ions on the basis of the three schemes discussed in the following. It should be noted that according to the formed complexes of lanthanum and yttrium in aqueous and organic phases sequentially [La.NO3.(H2O)7]2+ and [Y.NO3.(H2O)4]+, [La.NO3.(D2EHPA)2] and [Y.(NO3)2.D2EHPA] that had been proved by Alizadeh and co-workers [43], by reason of exactly similar conditions, we applied these complexes hereafter for our simulations.
Scheme 1- This scheme is based on the implicit solution model, such that the bare metal ion is solved directly in the continuum solvent by the use of the COSMO solvation model.
Scheme 2- This scheme, recognized as the monomer cycle, is based on the explicit solvation model, similar to what Dolg et al. mentioned [51]. For the monomer cycle, the ionic solute reacts with n separated water molecules. The gas phase metal ion is solvated in the first solvation sphere water molecules as below:
$${[La.N{O}_{3}]}_{\left(g\right)}^{2+}+7{\left[{H}_{2}O\right]}_{\left(aq\right)}=[La.N{O}_{3}.{\left({H}_{2}O\right)}_{7}{]}_{\left(aq\right)}^{2+}$$
15
$${[Y.(N{O}_{3}{)}_{2}]}_{\left(g\right)}^{+}+4{\left[{H}_{2}O\right]}_{\left(aq\right)}=[Y.(N{O}_{3}{)}_{2}.{\left({H}_{2}O\right)}_{4}{]}_{\left(aq\right)}^{+}$$
16
Scheme 3- The cluster method applied by Goddard et al. is used in the cluster cycle, such that, unlike the monomer cycle, a cluster of n water molecules reacts with the ionic solute in the cluster cycle. These clusters are used for geometry optimization and total energy computation [50]:
$${[La.N{O}_{3}]}_{\left(g\right)}^{2+}+[{H}_{2}O{]}_{{7}_{\left(aq\right)}}=[La.N{O}_{3}.{\left({H}_{2}O\right)}_{7}{]}_{\left(aq\right)}^{2+}$$
17
$${[Y.(N{O}_{3}{)}_{2}]}_{\left(g\right)}^{+}+[{H}_{2}O{]}_{{4}_{\left(aq\right)}}=[Y.(N{O}_{3}{)}_{2}.{\left({H}_{2}O\right)}_{4}{]}_{\left(aq\right)}^{+}$$
18
Table 2 presents the hydration energy (HE) of every metal in each scheme. This value is estimated by the energy difference between the ultimate hydrated complex and the sum of the energies of the water molecules and metal ionic complex.
Table 2
Calculated values of hydration energy (kcal/mol) and reaction free energy (kcal/mol) of La(III) and Y(III) ions
| Energy | Hydration of La ions | Hydration of Y ions |
| Scheme 1 | Scheme 2 | Scheme 3 | Scheme 1 | Scheme 2 | Scheme 3 |
| HE (kcal/mol) | -265.74 | -296.10 | -383.70 | -96.58 | -137.98 | -171.83 |
| ∆G (kcal/mol) | 0.40 | 75.18 | 16.30 | 0.09 | 42.03 | 16.70 |
| ∆∆G (kcal/mol) | -0.31 | -33.15 | 0.40 | | | |
As these values illuminate, according to all three schemes, the hydration energy for the formation of lanthanum hydrated complex is more than the rate for the yttrium hydrated complex. Thus, we can conclude that the stability of the lanthanum ionic complex is more than that of the yttrium ionic complex in the aqueous phase, and a more stable species of lanthanum is formed in the aqueous solution compared to yttrium. Furthermore, the analysis of free energy among three schemes shows that the results obtained from the free energy difference (∆∆G = 0.4 kcal/mol) of scheme 3 approximate to what is observed in reality compared to similar values in the other schemes. In reality, it is observed that the lanthanum complex further tends to be present in the aqueous solution compared to yttrium. Hence, the selectivity of water solvent is higher for solvating lanthanum complexes than those of yttrium.
5.3. Extraction Selectivity
Similar to what has been mentioned in some references, this study also showed that the binding energy of the gas phase was not a suitable tool for showing the higher experimental selectivity of Y(III) ions than that of La(III) ions towards the D2EHPA ligand. The reason for this issue is the ignorance of considering the solvent effect in both aqueous and organic phases. Metal ions should be extracted from the aqueous environment, wherein they severely take hydrated forms. Hence, it is necessary to calculate the solvation energy of lanthanum and yttrium ions in an aqueous environment to achieve an accurate estimation of extraction energy. To correctly estimate the manner of extraction selectivity and complexation studies in solvent extraction processes, we can employ thermodynamic calculations. As Fig. 2 illustrates, Born-Haber thermodynamic cycle is used for computing the solvent phase free energy of complexations [52].
Figure 2. Thermodynamic cycle for the calculation of extraction free energy (M = La(III) or Y(III) ion, L = D2EHPA−, and x = 1 and 2 for La(III) and Y(III), respectively)
Complexation selectivity for metal ions can be modeled by use of the extraction reaction presented below :
$${(M.xN{O}_{3})}_{\left(aq\right)}^{\left(3-x\right)+}+m{L}_{\left(org\right)}\to (M.xN{O}_{3}.mL{)}_{\left(org\right)}$$
19
In the following, the free energy of the above reaction complexation (∆Gext) is estimated by the thermodynamic cycle method, being highly successful in estimating the solution phase selectivity.
| \({\varDelta G}_{ext}={\varDelta G}_{g}+\varDelta {\varDelta G}_{sol}\) | (20) |
| \({\varDelta G}_{g}={G}_{{(M.xN{O}_{3}.mL)}_{\left(g\right)}}-\left({G}_{{(M.xN{O}_{3})}_{\left(g\right)}^{\left(3-x\right)+}}+m{G}_{{L}_{\left(g\right)}}\right)\) | (21) |
| \(\varDelta \varDelta {G}_{sol}=\varDelta {G}_{sol[M.xN{O}_{3}.mL]}-\left(\varDelta {G}_{sol\left[\right(M.xN{O}_{3}{)}^{\left(3-x\right)+}]}+m\varDelta {G}_{sol\left[L\right]}\right)\) | (22) |
∆Gg is calculated by the use of the free energy relevant to the structures optimized in the gas phase, and other terms associated with ∆Gsol in the solvent phase are computed by the use of these structures in the gas phase. The solvation energy rate of La(III) and Y(III) ions in the aqueous phase is determined by the cluster solvation model at this stage of computations. The selectivity of the D2EHPA ligand for the rare earth metal ions can be explained as the exchange reaction of metal cation, described below:
$$[Y.(N{O}_{3}{)}_{2}.L{]}_{\left(org\right)}+[La.N{O}_{3}{]}_{\left(aq\right)}^{2+}+{L}_{\left(org\right)}^{-}\to [Y.(N{O}_{3}{)}_{2}{]}_{\left(aq\right)}+[La.N{O}_{3}.2L{]}_{\left(org\right)}$$
23
Hence, selectivity can be explained by the difference in the free energy of extraction energy (∆∆Gext) as shown below (52–53):
$$\varDelta \varDelta {G}_{ext}={\varDelta G}_{[La.N{O}_{3}{]}^{2+}}-{\varDelta G}_{[Y.(N{O}_{3}{)}_{2}{]}^{+}}$$
24
Table 3 shows the values of the calculated free energies. A positive value of ∆∆Gext (+ 10.9 kcal/mol) indicates the selectivity of Y(III) ions to the D2EHPA ligand is more than that of La(III) ions. Thus, the selectivity process observed in the experimental studies was also confirmed in calculations. Compared to the energy of the metal ionic solvation, the solvation energy rate of the ligand and ligand-metal ion is very small. Hence, we can claim that aqueous solvation energy plays a paramount role in determining the selectivity process.
Table 3
Calculated free energy (kcal/mol) for extracting of La(III) and Y(III) with D2EHPA ligand at BLYP level of theory
| Reaction | ∆Gg | ∆∆Gsol | ∆Gext | ∆∆Gext |
| \((La.N{O}_{3}{)}_{\left(aq\right)}^{2+}+2{L}_{\left(org\right)}^{-}\to (La.N{O}_{3}.2L{)}_{\left(org\right)}\) | 22.9 | -17.3 | 5.6 | 10.9 |
| \((Y.(N{O}_{3}{)}_{2}{)}_{\left(aq\right)}^{+}+{L}_{\left(org\right)}^{-}\to (Y.(N{O}_{3}{)}_{2}.L{)}_{\left(org\right)}\) | 16.47 | -21.77 | -5.3 | |
It should be reminded that the thermodynamic parameters obtained by Van’t Hoff methods may not ideally be rational due to experimental uncertainties (54). Therefore, there may sometimes be contradictions between the results obtained by Van’t Hoff methods and computer computations. However, with respect to the results presented by both experimental studies (∆Gexp) and quantum calculations (∆Gext), there is an excellent correlation among the results of this study. The selectivity trend observed in the laboratory concerning rare earth elements extraction by the D2EHPA extractant can depend on different factors, including a difference in the ionic potential, ionic radius, the interaction of these elements with counter ions in the aqueous solution, and the hydration of the aqueous complexes of rare earth elements. However, regarding the theoretical computations of this study, it seems that the difference in the hydration of rare earth element complexes can be a key factor in their extraction selectivity with the D2EHPA extractant.
5.4. Bonding Analysis and Reactivity
When the interaction energy of the extraction reactions was calculated regardless of the solvent effect in the gas phase, it was observed that the value of this energy was larger for La(III) (-452.68 kcal/mol) than that of Y(III) (-192.35 kcal/mol). This trend counters the extractability trend observed in the extraction free energy calculations. To better perceive the reason for this event and better comprehend the metal-ligand complexes bonding, we studied the energy of the frontier molecular orbitals (LUMOs and HOMOs) and charge transfer on metal ions in the relevant complexes using population analysis. Table 4 displays the Mulliken charge and occupied orbitals for the related complexes. The charges of lanthanum and yttrium ions equal + 3 in the free state. It can be seen that the positive charge of lanthanum and yttrium ions decreases in the formed aqueous complex. This issue shows that electrons are transferred from the existing nitrate ligand in the aqueous solution to the lanthanum and yttrium ions and form coordination bonds. Due to the transfer of more electrons because of the further coordination of the nitrate ions with yttrium (two coordination positions), the partial charge of yttrium (+ 1.86) experiences more reduction compared to the free state and the partial charge of Lanthanum (+ 1.91).
Table 4
Calculated values of Mulliken charges (Q) and occupied orbitals (s, p, d, and f) at BLYP level of theory
| Complex | s | p | d | f | Q |
| [LaNO3.(H2O)7]2+ | 12.2 | 26.8 | 29.9 | 14.1 | 1.91 |
| [LaNO3.(D2EHPA)2] | 12.1 | 26.5 | 30.1 | 14 | 1.65 |
| [Y(NO3)2.(H2O)4]+ | 10.4 | 24.7 | 24.6 | 0 | 1.86 |
| [Y(NO3)2.D2EHPA] | 10.5 | 23.4 | 22.9 | 0 | 1.69 |
The charge transfer is larger for La(III) than that of Y(III) in their complexes extracted by the D2EHPA ligand. This trend is consistent with the lower interaction energy of Y(III) than that of La(III) in the gas phase. Furthermore, the value of the Mulliken charge on the atom center of La(III) is larger than this value for the Y(III) atom center. This denotes the lower covalent character of La(III) complex than that of Y(III). In the DFT studies, no treatment was applied to electrons, and all electrons were considered as ground state valence in calculations. The crowdedness of electrons in the s, p, and d orbitals of the lanthanum and yttrium ions reflects the covalent nature of bonding after complexations in the aqueous nitrate medium and complexations with D2EHPA.
The energy levels of HOMO and LUMO, recognized as frontier molecular orbitals and the reactivity controllers of molecular complexation, were also estimated for the hydrated complexes of these metals. LUMO modifies the ability of the molecule for accepting electrons in any structure, while HOMO shows the electron-donating ability of the molecule. The difference between the energy values of HOMO and LUMO is called the HOMO-LUMO energy gap, which shows the probable charge transfer interaction that occurred within a molecule. Molecules with a small energy gap are generally accompanied by high chemical reactivity and low kinetic stability besides being considered as soft molecules. However, molecules with a large energy gap possess higher stability and are recognized as hard molecules. It is because they oppose their charge transfer, distribution changes, and electron density. In Table 5, some chemical properties of the studied structures were computed by the use of DFT studies at the BLYP level of theory. Concerning the difference in the number of nitrate ligands in the first coordination clusters of the lanthanum and yttrium, the amount of the energy released due to electron addition to the aqueous complex of these elements also differs. The electron affinity amount for the aqueous complex of yttrium (0.24 eV) is smaller than that of lanthanum (0.43 eV) due to the addition of the second electron to the hydrated complex. Hence, the value of the electron affinity is larger for the hydrated complex of lanthanum owing to the addition of a single electron to its structure.
Table 5
Calculated chemical parameters for studying of the reactivity of the complexes
| Complex | IP (eV) | EA (eV) | X (eV) | η (eV) | S (eV) |
| [LaNO3.(H2O)7]2+ | 0.48 | 0.43 | 0.46 | 0.02 | 42.18 |
| [LaNO3.(D2EHPA)2] | 0.24 | 0.14 | 0.19 | 0.05 | 20.85 |
| [Y(NO3)2.(H2O)4]+ | 0.39 | 0.24 | 0.31 | 0.07 | 13.77 |
| [Y(NO3)2.D2EHPA] | 0.26 | 0.13 | 0.20 | 0.06 | 15.60 |
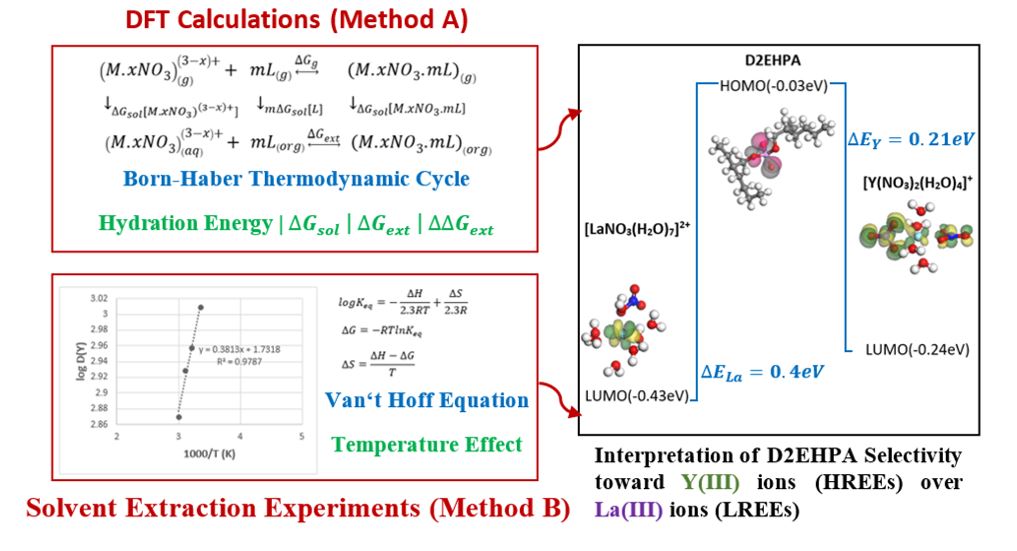
As observed, the hardness of yttrium hydrated complex (0.072 eV) is more than that of the lanthanum hydrated complex (0.024 eV). This means that, in the presence of an electric field, the electron cloud of the yttrium-containing molecule is less distorted than that of lanthanum. The chemical softness of the lanthanum complex extracted by D2EHPA (20.85 eV) is higher than that of the yttrium complex extracted similarly (15.6 eV). This indicates that lanthanum forms a more unstable complex in the organic phase compared to yttrium. Therefore, we can conclude that yttrium forms a stronger bond with the D2EHPA ligand than that of lanthanum. This issue can be reckoned as one of the reasons for the experimental observations respecting the harder chemical conditions needed for yttrium stripping for the transfer of ions from the organic to the aqueous phase compared to the lanthanum stripping. Likewise, the larger ionization potential of the extracted lanthanum complex (0.24 eV) denotes that the energy rate needed for removing an electron from yttrium is more than the rate needed for lanthanum. That is why we can claim that yttrium forms a more stable complex in the organic phase, and it is more difficult to omit the D2EHPA ligand, as an electron donor, from the extracted structure. Furthermore, the higher electronegativity of the lanthanum aqueous complex (0.46 eV) than that of the yttrium aqueous complex (0.31 eV) shows the further capability of this molecule in attracting the electron towards itself during bond formation. Figure 3 illustrates the levels of the frontier molecular orbitals and their related shapes for the considered combinations.
Figure 3. Shapes of frontier molecular orbitals for lanthanum complexes, yttrium complexes, and D2EHPA extractant at BLYP level of theory
As observed, the D2EHPA ligand contributes to the HOMO shapes of the extracted complexes of rare earth elements, and lanthanum and yttrium contribute to the LUMO shapes of those complexes. For the formation of a bond between D2EHPA and every one of the yttrium and lanthanum ionic complexes, an electron should move from the highest occupied molecular orbital ; i.e., the HOMO related to the D2EHPA ligand, to the lowest unoccupied molecular orbital; i.e., the LUMO related to every metal ionic complexes. For this reason, as the energy level difference of these orbitals (∆Eorbital) becomes smaller, the electron transfer is simpler, and the reactivity is higher. This difference in the energy level equals 0.4 eV and 0.21 eV for the bonding of D2EHPA with the lanthanum and yttrium ionic complexes, respectively. Hence, this result implies that the formation of a bond between D2EHPA and the yttrium ionic complex is simpler than that of the lanthanum ionic complex. In this condition, a more stable complex is formed. Table 6 shows the Mayer bond order analysis at the BLYP level of theory and its results.
Table 6
Mayer bond orders of D2EHPA molecule after the formation of extracted complexes of La(III) and Y(III)
| Complex | Bonds | Mayer Bond Order |
| [LaNO3.(D2EHPA)2] | P = O | 1.25 |
| P-O− | 1.23 |
| [Y(NO3)2.D2EHPA] | P = O | 1.14 |
| P-O− | 1.19 |
Compared to La(III), the smaller values of the bond order for the electron donor head group in the D2EHPA ligand in the extracted Y(III) complex show the weakening of these bonds due to the formation of a stronger bond of the ligand with the relevant metal. This issue qualitatively interprets the selectivity and stronger bond of the yttrium metal with the extractant ligand compared to lanthanum. Besides, the partial charge distribution shows that oxygen atoms have a higher negative charge in D2EHPA. Since the electron density of the double bond oxygen atom is higher than that of the single bond oxygen atom connected to the phosphor atom in the D2EHPA structure, and the single bond oxygen atom is a weaker donor atom, both lanthanum and yttrium form a chelating bond with this extractant.
{kind=link}