Electrochemistry
Electrochemical studies of prepared monomers 1 and 2 showed they were electroactive and underwent multiple redox processes, but reversible reduction processes could not be definitively stated due to low concentration of monomer in the solution (28 µM and 23 µM, respectively for 1 and 2). However, it was possible to conduct the appropriate studies on the anodic and anodic-cathodic waveforms, in which the electro-oxidation reaction was used to obtain layers of electroactive materials.
The study found that the oxidation of the monomers was linked to the oxidation of the perimidine unit, which is described in previous studies as coupling radical cations. The oxidation of monomers 1 and 2 was irreversible with max peaks at + 0.93V and + 0.86V, respectively. The products of the electro-oxidation were different depending on the electrode cycle, leading to varying chemical structures and electroactivity. Table S1 shows the potentials obtained in the anode and anode-cathode cycles and Fig. S3 shows the potential ranges of the three main fractions of products.
The oxidation of the studied monomers was related to the oxidation of the perimidine unit, which is described in previous studies as coupling radical cations22–25. Also, the prepared monomers displayed irreversible oxidation with the max. at + 0.93 V for monomer 1 and + 0.86 V for monomer 2 (Fig. S2). Reports have shown that depending on the position of two perimidine units during electrocoupling, various fractions of products could be characterized by different electroactivity depending on the potential. The type of products can differ depending on the deposition in an anode or anode-cathode cycle due to the distinct organization of molecules on the electrode during the cathodic cycle, which involves n-type doping and can rearrange the material’s structure26. Tab. S1 shows the potentials of materials obtained during the electro-oxidation of monomers 1 and 2 in the anode and anode-cathode cycles. In the anodic waveform, four fractions of products were observed for polymer 1 (p1), and three fractions for polymer 2 (p2). The products were divided into three main fractions. Fig. S3 shows the potential ranges of the fractions, from − 0.25 to 0.19 V for 1ox, 0.20 to 0.64 V for 2ox, and 0.65 to 1.15 V for 3ox; which were products of p1 and p2.
CA was employed at the monomer-oxidizing potentials to determine that the formation of 2ox fraction required an alternating potential. Fig. S4 shows that at a constant potential, the current intensity for the product polarization was comparable to the background (Fig. S4c), hence, 2ox product was not formed. However, during the electrodeposition process, by sweeping the potential from 0.9 to 0.7 V, 2ox fraction was obtained (Fig. S4d). Therefore, 2ox fraction was a mixture of decoupled products, in which its preparation required a lower potential after reaching the monomer’s oxidation peak.
During the electro-oxidation of the anodic-cathodic waveform, a widening of the current response and two reduction peaks for each of the material was observed (for p1 at -1.52 and − 2.00 V; for p2 at -0.98 and − 1.44 V). These are marked in Fig. 1 as red1 and red2. The presence of these two reduction peaks proved that the redox couples derived from the monomers were maintained (Fig. S2). The reduction of both materials and monomers was comparable to the electroactivity of BBL polymer27, which consists of isomeric units of compound 1. The redox process in BBL is reversible and contains two single electron transfer steps at the potentials of -1.14 and − 1.48 V vs. Fc/Fc+ 28. A two-stage reduction was also observed with pyrrolo[3,4-m]phthaloperinedione (for p1) and [3, 8]phenanthrolinedione (for p2) cores. 1red and 2red peaks that corresponded to these processes were broadened due to the development of various types of products and non-covalent interactions between the material segments in the solid.
Sweeping the potential in a wide range (Fig. S5) did not degrade or dissolve the materials, which proved their stability. During the anodic-cathodic waveform, materials with higher current responses were always obtained that did not disappear in the subsequent sweep cycles, hence, the electropolymerization method, which included the potential sweep in the reduction region, was better. In addition, an increased current intensity was detected compared to the background for the obtained materials. This suggested the conductivity of the materials over the entire range of the potential sweep. In the case of p2, the boundary between the reduction and oxidation regions was invisible (Fig. S3), which was considered as the electrochemical energy gap of the material close or equal to zero. The same gap for p1 was equal to 0.44 V. Main issues arose during the development of further structural studies due to the low concentration of monomers 1 and 2, making it impossible to obtain a sufficient amount of p1 and p2 materials to peruse further research techniques. Therefore, to explain the structure of the obtained materials, we decided to use quantum chemical calculations, which will be presented later in the article.
Quantum chemical calculations
Quantum chemical calculations were utilized to determine the energies and localization of molecular orbitals for the syn- and anti-isomers present in monomers 1 and 2. Density Functional Theory (DFT) was employed to obtain information regarding the expected redox potentials and the preferred reactivity sites of these systems (Tab. S2). These calculations were supplemented with localizations of spin densities for the appropriate radical ionic states (Tab. S3). HOMO (Highest Occupied Molecular Orbital) was located mainly on the perimidine units for all calculated monomers and the energies values were − 5.33 eV for monomer 1 and − 5.11 eV for monomer 2. The energy and localization of orbitals are the same for syn- and anti-isomers, meaning their properties should be similar during experiments. The energy value of HOMO-1 was lower than that of HOMO (-5.47 eV for monomer 1 and − 5.40 eV for monomer 2). This suggested the ‘electronic communication’ of the two terminal perimidine units by aromatic cores. Additionally, it led to partial delocalization of the orbitals and extension of the charge throughout the molecule. LUMO (Lowest Unoccupied Molecular Orbital) was located mainly on the aromatic core units (pyrrolo[3,4-m]phthaloperinedione and [3, 8]phenanthrolinedione) and partially delocalized towards the oxygens in the molecule. The energy values of these orbitals were − 2.94 eV for monomer 1 and − 3.06 eV for monomer 2.
Based on the calculated localization of electron spin density for excited forms, we proposed the position of the covalent connection in the electro-oxidation products. All calculated radical cations showed that the spin was located mainly on the four carbon positions 1, 3, 4, and 6 and nitrogen atoms (Tab. S3). The entire spin of both radical cations and diradical dications was located on the perimidine units. Hence, the spin of the radical anions and diradical dianions was located on the aromatic central unit e.g., pyrrolo[3,4-m]phthaloperinedione and [3, 8]phenanthrolinedione.
To facilitate the calculations, some assumptions and simplifications were used. The syn- and anti-isomers of monomer 2 were only calculated. Three main groups of products were categorized (Fig. 2) - protonated bis-perimidine (Tab. S4), semi-ladder bis-perimidine (Tab. S5); and ladder bis-perimidine (Tab. S6). Additional mers (n = 4 or 8) were computed with only 4 possible structures of ladder bis-perimidine products (Tab. S8 and S9). Intermolecular interactions and supporting electrolyte ions were ignored. The calculated HOMO value for monomer 2 was − 5.11 eV, which was compared with the calculated energies of the orbitals for electro-oxidation products. Additionally, a difference of 1 eV corresponded to a difference of 1 V for CV and DPV measurements29.
Based on the calculations performed for the possible electro-oxidation products, we proposed a series of products obtained during the electropolymerization of monomer 2 (Fig. 3). Firstly, during the recombination of perimidine radical cations, the simplest connection was the protonated bond in the dication segment, which was previously described as 1ox fraction. The calculated LUMO value of this type of connection was between − 4.44 and − 4.65 eV. The oxidation potential of monomer 2 was 0.84 V, so the calculated values for the products of fraction 1ox should have a potential of 0.17 V. The electrochemical response for fraction 1ox was in the range of -0.2 to 0.2 V for DPV measurement and it is most probably a set of redox reactions for bis-perimidine junctions in the form of dications. During the previous calculations of bis-perimidine dications, it was found that some bonds, such as 1,1’ or 1,6’, were more stable due to the possibility of possessing hydrogen bond interactions inside the molecule. The products stabilized in this way did not undergo any further oxidation reaction23,25.
2ox and 3ox fractions were products of deprotonation of bis-perimidine dications to neutral bonds of semi-ladder and ladder bis-perimidines, respectively. According to the calculations, these fractions should be related to 2ox and 3ox at potentials 0.36 and 0.91 V, respectively. In this case, the calculations were consistent with DPV measurement results. To test our theory of the generation of ladder connections in 2ox fraction, electrochemical analysis of perylene was conducted due to ladder dimers being derivatives of perylene (Fig. S6). The oxidation potential of perylene was 0.55 eV, which confirmed the presence of a perylene unit in p2 material, found in 2ox fraction. Additionally, the current response assigned to 3ox may be caused by the oxidation of perimidine terminal units and monomers included in the material structure.
In the reduction region (below 0 V) current responses on DPV curve starting from − 0.4 V were observed. These were generated during the reduction of neutral dimers and larger oligomers with semi-ladder and ladder perimidines. During the calculations, it was found that the reduction of longer oligomers should occur at higher potentials compared to the monomer, owing to the energy value of LUMO orbital larger oligomers becoming lower. Additional signals below − 1 V originated from the reduction of the monomer occluded in the polymer structure, the terminal units, and from further stages of the reduction of the oligomers.
The calculations showed that with each additional monomer unit in the polymer chain, the band gap decreases to a limit of 0.93 eV, as shown by fitting the trend function for the calculated oligomers (Fig. S7). The geometry and values of the boundary orbitals were calculated in a pure solvent, ignoring intermolecular interactions.
Also, calculations for neutral ladder oligomers of 2, 4, and 8 monomer units were performed. The calculations showed that with each additional monomer unit in the polymer chain, the band gap decreases to a limit of 0.93 eV, as shown by fitting the trend function for the calculated oligomers (Fig. S7). The geometry and values of the boundary orbitals were calculated in a pure solvent, ignoring intermolecular interactions. In a condensed system, hydrogen or π-electron interactions (such as π-stacking) may occur, which stabilized the structure and reduced the band gap of the material30–32.
Mechanism of electropolymerization
p1 and p2 polymers were produced via electropolymerization of perimidine units. These materials arose due to the recombination reactions of perimidine radical cations, generated by the application of an oxidizing potential. As shown above, the spin density of radical cations was concentrated at positions 1, 3, 4, and 6. Hence, bonding at these perimidine positions was possible. The bond at position 1 was in a non-protonated form due to the stabilization of the proton with the nearby amide group. Binding through the remaining positions stemmed from the formation of deprotonated bis-perimidine segment first (semi-ladder segments). In the subsequent oxidation cycles, a certain amount of semi-ladder segments could condense into a ladder, which is especially favored by the planarity of the monomer and product structure (Fig. 3).
The deposition reactions of p1 and p2 are shown in Fig. S8 and Fig. S9, respectively. In the anodic-cathodic wave, p1 showed an additional orientation of the molecules after the application of low potential and the formation of π-stacking interactions that stabilized the polymer structure. In the case of p2, deposition in an anodic-cathodic waveform promoted the radical anions to react upon the application of reducing potentials. The correct orientation of the oligomer segments caused the formation of covalent bonds between the naphthalene units, resulting in the stiffening of the polymer structure and increased stability of the material during cyclic polarization, compared to p1. This phenomenon was previously observed for isoindole isomers containing a perimidine unit17.
Electrochemical properties of p2 material
Notably, p1 material was not stable during prolonged polarization at both reducing and oxidizing potentials. Its low stability, compared to p2, was possibly due to the considerably smaller amount of interactions between planar pyrrolo[3,4-m]phthaloperinedione central units. In addition, p2 during the reduction process formed covalent bonds between the [3, 8]phenanthrolinedione cores. Therefore, we will present in this work further research only for p2 material.
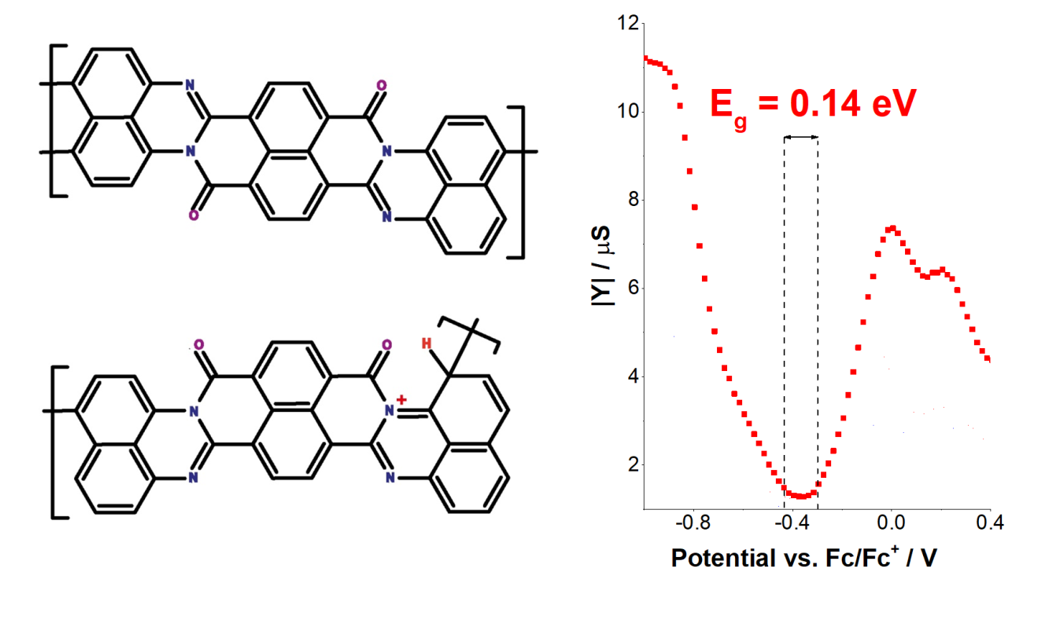
The bandgap value for p2 material was impossible to determine using CV Cardona’s method34, because, according to calculations, it had an energy gap below zero (Fig. S10). By measuring the admittance of p2 material, the electrochemical band gap of the material deposited during 10, 20, and 30 cycles were determined (Fig. 4). The admittance measurement revealed that with an increasing number of deposition cycles, the value of LUMO orbital energy for the material decreased, while HOMO energy remained unchanged. The decreasing LUMO value with each deposition cycle tightened the band gap to the value of 0.14 eV for the 30th cycle layer.
To gain a better understanding of the doping behavior, p2 material was studied using electrochemical impedance spectroscopy (EIS) for 10 and 20 cycles of deposition. This technique allows for the analysis of complex electrode processes and has previously been used to describe polarization-induced ion antiport35 as well as the determination of the conductivity type of semiconducting materials36. We proposed an equivalent circuit describing the polymer film under polarization, which is shown in Fig. 5. The circuit was not processable due to an infinite number of parameters, hence a simplified approach was employed. The circuit described charge relaxation, meaning the propagation of the charge from the polymer-electrode to the polymer-solution interface. Each R-C branch stood for a charge transfer process, which was defined by its rate and sensitivity to the potential change.
The introduction of a time-dependent element in the equivalent circuit for impedance measurement allowed for the characterization of p2 films for 10 and 20 deposition cycles (Fig. 6). The processes that occur in the thin and thick films were the same, but observation of the processes was much more pronounced in the case of the thicker film (20 cycles of deposition). The fastest processes characterized by a time-constant below 50 µs (Fig. 6, b fast) were not present in this material. However, processes for a medium and slow time-constant above 0.1 ms were detected. The impedance measurements revealed that two electrochemical processes occur in p2 polymer with a time-constant above 10 ms. These processes were caused by the dedoping of the polymer, i.e., the migration of counter ions from the material to the electrolyte due to the changed polarization of the working electrode. They occurred at potentials of c.a. -0.75 and 0.3 V (Fig. 6b slow). Other processes were considered as oxidation or reduction of individual segments of the material, which was accompanied by the migration of electrolyte ions into the material (doping). Therefore, p2 was mainly a redox polymer. However, high charging currents of the material, especially in the range from − 0.6 to -0.2 V, indicated conductivity along the polymer chain and intermolecular conductivity. Most likely, the conductivity in this range increased due to trapped counter ions in the material’s structure.
{kind=link}