This is the first study to demonstrate that downregulation of A2A AR and A2B AR contributes to the development of fibrotic diseases, and that fibrotic progression can be inhibited by upregulating A2A AR and A2B AR. Downregulation of A2A AR and A2B AR and a reduction in Epac levels were observed in murine models of fibrotic liver and lung diseases, suggesting that downregulation of A2A AR and A2B AR contribute to the development of fibrotic diseases in the liver and lung by attenuating cAMP signaling. MF treatment upregulated A2A AR and A2B AR, elevated Epac levels, and inhibited inflammatory and fibrotic responses in murine models of fibrotic diseases, suggesting that the upregulation of A2A AR and A2B AR inhibits inflammatory and fibrotic processes by enhancing cAMP signaling. Therefore, A2A AR and A2B AR may be therapeutic targets against inflammatory and fibrotic diseases, including liver and lung fibrosis, and A2A AR and A2B AR-upregulating agents, such as MF, are potential therapeutic agents for fibrotic diseases.
Cyclic cAMP modulates inflammatory and fibrotic responses via Epac- and PKA-mediated pathways. Cyclic AMP/Epac signaling exerts anti-fibrotic actions by inhibiting epithelial–mesenchymal transformation and ECM formation. In contrast, pro-fibrotic agents, such as TGFβ and angiotensin II, downregulate Epac, thereby promoting fibrotic responses [9]. In addition, cAMP/PKA signaling can inhibit inflammatory and fibrotic responses. Continuously elevated cAMP levels inhibit immune cell functions, such as the proliferation and activation of T and B cells [8], and PKA signaling may be involved in anti-inflammatory and anti-fibrotic pathways [8, 24]. Downregulation of KCa2.3 and KCa3.1 may play a critical role in the anti-inflammatory and anti-fibrotic effects of cAMP. Pro-inflammatory cytokines and pro-fibrotic growth factors, such as TGFβ [12] and VEGF [25], upregulate KCa3.1 and KCa2.3, and pharmacological inhibition or knockdown of KCa3.1 and KCa2.3 inhibits in vitro and in vivo fibrotic responses [12, 13, 26, 27]. Cyclic AMP was found to reduce the expression of KCa3.1 and KCa2.3 via an Epac-mediated pathway [12], and inhibited the KCa3.1 current via PKA-dependent phosphorylation of KCa3.1 [14]. MF may exert anti-fibrotic and anti-fibrotic effects via cAMP/Epac and PKA/KCa2.3 and KCa3.1-mediated pathways in various organs, such as the liver and lungs [12].
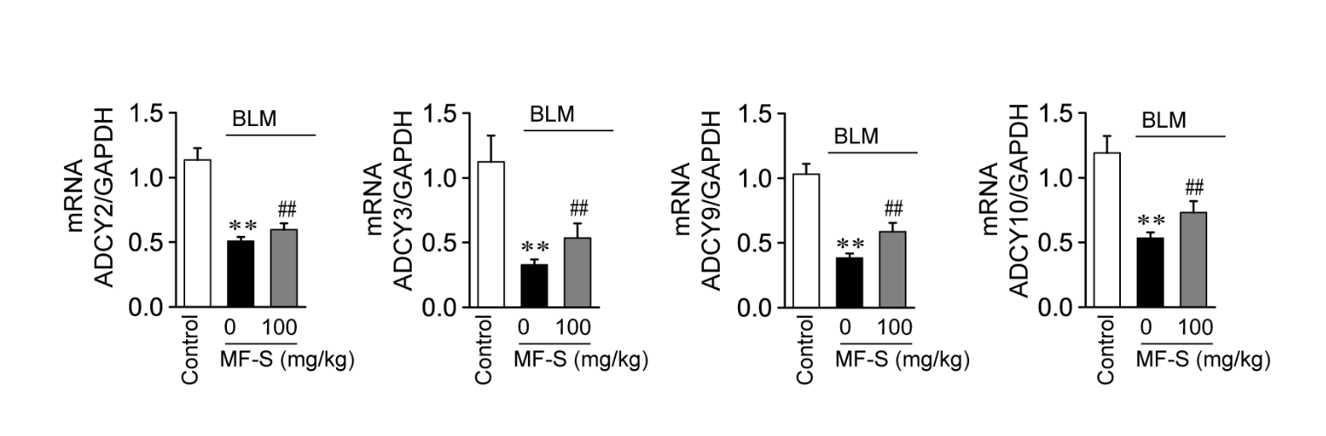
A2A AR and A2B AR activate cAMP signaling via the Gs protein, whereas A1 AR and A3 AR inhibit cAMP signaling via the Gi protein. Thus, the downregulation of A2A AR and A2B AR and upregulation of A1 AR and A3 AR, as observed in murine models of fibrotic diseases, indicate that cAMP signaling is suppressed. Suppression of cAMP signaling may contribute to the development of fibrotic diseases. In contrast, upregulation of A2A AR and A2B AR and downregulation of A1 AR and A3 AR, as observed in MF-treated murine models of fibrotic diseases, indicate that cAMP signaling is augmented by MF treatment in fibrotic models. Therefore, augmented cAMP signaling may attenuate inflammatory and fibrotic progression in fibrotic diseases. These results suggest that A2A AR and A2B AR play important roles in the inhibition of liver and pulmonary fibrosis.
A2A AR is distributed in the liver, lungs, and immune system and is expressed in immune cells and fibroblasts [16]. The A2A AR acts as an endogenous modulator of inflammation and tissue repair. Expression levels of A2A AR were markedly decreased in lung tissue from a murine model of BLM-induced pulmonary fibrosis (Fig. 1), which is consistent with the finding that A2A AR was downregulated in patients with severe IPF [28]. In addition, A2A AR-null mice were more susceptible to BLM-induced lung injuries [17]. These results suggest that A2A AR is a potential target for inflammatory and fibrotic diseases.
The role of A2B AR in the inflammatory response remains controversial. A2B AR is distributed in the bowels and lungs and expressed on immune cells [16]; in addition, A2B AR has both pro-inflammatory and anti-inflammatory effects. Pharmacological inhibition or knockout of A2B AR suppresses intestinal inflammation in murine colitis models [29] and exacerbates inflammation of dextran sodium sulfate colitis in mice [30]. Anti-inflammatory effects are generated by coupling with protein Gs, while proinflammatory effects are generated by coupling with protein Gq [19]. However, A2B AR may be a potential target for treating acute lung injuries [31]. Pharmacological inhibition or deletion of the A2B AR enhanced pulmonary inflammation, and an A2B AR agonist attenuated pulmonary inflammation.
It is unclear how MF restores downregulated A2A AR and A2B AR in murine models of fibrotic diseases and TGFβ-treated NHLFs, and how MF blocks TGFβ-induced alterations in KCa2.3, KCa3.1, Col1, and α-SMA. It is known that MF increases cAMP levels in various cells, and cAMP regulates the gene expression of various molecules. As shown by the cAMP regulation of KCa2.3 and KCa3.1 expression, MF may regulate the expression levels of these proteins by enhancing cAMP signaling. Cyclic AMP-induced KCa2.3 and KCa3.1 downregulation may inhibit Col1 and α-SMA production by attenuating Ca2+ signaling. However, further studies are necessary to clarify the mechanism by which MF regulates AR expression.
Chronic persistent inflammation and epithelial mesenchymal transition (EMT) are two important pathogenic events of fibrotic diseases, including IPF [32]. Therefore, the inhibition of inflammation or EMT may be an efficient therapeutic strategy for fibrotic diseases. As observed in the present study, downregulation of A2A AR and A2B AR contributed to the development of fibrotic diseases in the liver and lungs, whereas upregulation of A2A AR and A2B AR inhibited inflammatory and fibrotic responses. The activation of inflammatory and fibrotic processes, as evidenced by reduced Epac signaling and α-SMA upregulation, was inhibited by the upregulation of A2A AR and A2B AR. These results strongly suggest that A2A AR- and A2B AR-upregulating agents, such as MF, may serve as novel therapies to inhibit inflammatory and fibrotic progression via cAMP-mediated pathways.
{kind=link}