Adipose derived stem cells (ADSCs) have the potential to attenuate osteoarthritis (OA); however, complications such as immune rejection and tumour formation limit their application. Exosomes (Exos)-mediated acellular therapy is promising in alleviating OA. This study aims to confirm whether ADSC-exos derived from infrapatellar fat pad (IPFP, ExosIPFP) are more suitable for ameliorating OA than ADSC-exos derived from subcutaneous fat (ScAT, ExoScAT) in vitro and in DMM models. Then, we investigated the regulatory mechanism by which the two kinds of Exos inhibit extracellular matrix (ECM) degradation in OA. ADSCs were successfully isolated and Exos were then obtained. ExosIPFP exhibited better attenuated effects on osteoarthritic chondrocytes in vitro and in vivo than ExoScAT. Small RNA sequencing was performed and the results shown that miR-99b-3p was upregulated in ExosIPFP. In vitro experiments confirmed that ADAMTS4 is a direct downstream target of miR-99b-3p. Over-expression miR-99b-3p in ExosScAT (ExosScAT-99b-3p) indicated that miR-99b-3p serves a positive role for OA treatment by inhibiting ADAMTS4 expression both in vitro and in vivo. In addition, hydrogel microparticles (HMPs) system was prepared by microfluidic technology, and confirmed the beneficial results for long-term therapeutic by continuous release of Exos. Take together, these results suggest that the therapeutic effects of ADSC-Exos may vary according to differential expression of miRNAs. Exosomal miR-99b-3p may act as a promising therapeutic strategy for OA, in addition, the injectable HMPs act as a sustained local drug release system, therefore representing great potential for treating OA and other diseases.
Research Article
Sustained released of microRNA-99b-3p abundant exosomes derived from adipose stem cell encapsulated with hydrogel microparticles (HMPs) for long-term osteoarthritis treatment
https://doi.org/10.21203/rs.3.rs-2621379/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
osteoarthritis
exosomes
microRNA
microfluidics
hydrogel microparticles
OA is a slowly progressive and degenerative disorder that induces irreversible pathological changes such as damage to articular cartilage, remodelling of subchondral bone, inflammation of the synovial membrane, degeneration of ligaments and changes in muscles.[1] Numerous factors contribute to the development of OA, such as body weight, diet, age, and history of trauma. The gradual loss of chondrocytes is the major cause of OA since chondrocytes are critical for the formation and maintenance of articular cartilage.[2] OA is characterized by the loss of ECM and cartilage destruction. [3, 4] Promoting articular cartilage repair or regeneration is the key to preventing OA progression. However, current treatments for OA are inadequate in terms of therapeutic effect.
Recently, mesenchymal stem cells (MSCs) isolated from human adipose tissue have been identified as an important source of multipotent adult stem cells, which have been widely used as an engineering stem cell source for tissue regeneration. [5–7] Adipose tissue is found in many parts of the human body, and the IPFP and ScAT are common tissues types for stem cell extraction. ADSCs have several advantages over other MSCs. Intra-articular injection of ADSCs improves the function and reduces pain and cartilage defects of the knee joint.[8] In addition, ADSCs can be more easily cultured and obtained in vastly greater quantities by less aggressive methods than other MSCs.[9]
Histologically, ScAT is mainly regarded as an endocrine and metabolic pool and has a mesodermal origin that is similar to bone and cartilage.[10] Moreover, it secretes a number of bioactive substances that exhibit anti-inflammatory and immunomodulatory characteristics.[11] IPFP is a mass of fibrous adipose tissue, mainly composed of synovium and subsynovial adipose tissue.[12] IPFP has more blood vessels and nerves and is more similar to visceral fat than ScAT.[13] IPFP-derived stem cells (IPFSCs) exhibit unique properties in terms of proliferation capacity and multilineage differentiation potential.[14] Some studies suggest that the therapeutic effect of IPFSCs on OA is better than that of subcutaneous adipose-derived stem cells (ScASCs). Mochizuki et al showed that IPFSCs have higher chondrogenic potential than ScASCs.[12]
However, the transplantation of MSCs also has risks that may cause immune rejection and potential tumorigenicity.[15] An increasing amount of evidence supports that the mechanisms underlying the therapeutic effects are likely paracrine mechanisms of MSCs, particularly exosome secretion.[16] Since the first report, MSC Exos have replicated many of the wide-ranging therapeutic effects of MSCs in numerous animal models of injuries and diseases. However, it is unknown which kind of ADSC exosome (ADSC-exo) is more effective in the treatment of OA.[15–17]
Exos are membranous vesicles with diameters ranging from 30 to 150 nm that move into the ECM after the integration of intracellular polyvesicles and cell membranes.[18] These vesicles can be secreted by many kinds of cells and have been reported to promote chondrocyte regeneration, inhibit apoptosis, and improve ECM balance, thus affecting the fate of cells or tissues.[18–21] Moreover, Exos contain many functional microRNAs (miRNAs), and miRNAs have the ability to modulate the expression of multiple genes implicated in various kinds of physiological functions and disease processes, such as OA. Previous studies have demonstrated that miRNAs play a key role in mediating the effects of the main risk factors for OA through the control of target genes and are critical in the pathological process of OA.[22–25]
Clinically, the IPFP is often routinely removed and disposed of as surgical waste during arthroscopy or open knee surgery, and most of it comes from elderly patients (who usually also have chronic diseases) with knee arthroplasty.[18, 26] Therefore, it has higher expression of inflammatory genes, releases more inflammatory factors, and contains more immune cells than ScAT.[27, 28] ScAT can be easily obtained from patients in large volumes with minimal inflammation by liposuction.[29] Therefore, it is particularly important to explore the difference in the expression of miRNAs between the two ADSC-derived exosome types (ExosScAT and ExosIPFP). By regulating the expression of key miRNAs, the effect of ExosScAT in OA treatment may be better than that of ExosIPFP. In future clinical work, abundant resources of ScAT will allow large quantities of stem cell culture and Exos to be produced to obtain enough high-quality exosomes to facilitate their use as effective biological agents for OA treatment.
Additionally, Exos have a short half-life in vivo, and injected Exos are easily cleared by the body’s immune system, resulting in a short retention time and low bioavailability at the target site, which limits their therapeutic potential in the clinic. Hydrogels can be used as effective drug loading materials to control drug release in situ. Various kinds of hydrogels have been used to protect Exos and enhance the therapeutic effects of Exos.[30–33] Among these hydrogel materials, poly(ethylene glycol) (PEG) has been extensively used for the fabrication of biocompatible hydrogels for tissue engineering and cartilage repair.[34, 35] Hyaluronic acid (HA) is one of the most versatile and fascinating macromolecules in the natural world and is one of the most widely used medical treatments for OA.[36] It can be modified with various functional moieties to form functional hydrogels. We have developed a kind of injectable hybrid hydrogel through hyperbranched poly(ethylene glycol) (HB-PEG) and thiolated HA (SHHA) crosslinking, and this hydrogel exhibited versatile properties, good biocompatibility, and ease of application.[37] However, due to the heterogeneous shape and large size of the hydrogel, high and uneven injection forces, which may damage healthy tissue and cause discomfort to the patients, are inevitable during the injection process.[38] Unlike conventional hydrogels, the spherical shape and relatively uniform size of HMPs significantly enhance injectability, and HMPs prepared by microfluidic methods have been widely used in various fields due to their enhanced structural stability, uniform size and good injectability.[39] In the presence of hyaluronidase in the knee joint, HA-based HMPs act as sustained release carriers that gradually degrade and release their contents.
In this study, we demonstrated the great importance of exosomal miRNAs in targeting cartilage and promoting ECM synthesis in OA. Moreover, we demonstrated the importance of the sustained release and prolonged effect of Exos for the treatment of OA. As shown in Scheme 1, we first studied whether ADSC-exos derived from IPFP are more suitable for ameliorating OA than ADSC-exos derived from ScAT in vitro and in a DMM model. Then, we investigated the regulatory mechanism by which the two kinds of ADSC-exo inhibit chondrocyte degeneration in OA. Through small RNA sequencing and bioinformatic analysis, we found that miR-99b-3p, which is more abundant in ExosIPFP, may alleviate the development of OA by suppressing the expression of ADAMTS4 and reducing the loss of ECM. Finally, to address the short half-life of Exos, we prepared HMPs via the Michael addition reaction between thiol-functionalized HA (SH-HA) and hyperbranched poly(ethylene glycol) diarylate (HB-PEGDA) in a droplet-based microfluidic device to yield Exos with abundant miR-99b-3p. The HMPs sustained the release of Exos for local and long-term therapeutic effects in the knee joint. In the mouse OA model, the injectable HMPs@ExomiR−99b−3p effectively alleviated OA progression by promoting ECM synthesis. Taken together, these results indicate that injectable HMPs with miRNA-abundant Exos that progressively target the ECM can be used for long-term therapy in OA and have great potential for application in future clinical work.
2.1. Ethics Statement.
Adult male Sprague-Dawley rats (6-8 weeks old, weighing 220 ± 20g) were purchased from the Animal Centre of Nanjing First Hospital, Nanjing medical university (Nanjing, China) and raised in specific pathogen-free animal cages under constant temperature and humidity and a 12h/12h dark/light cycle with adequate food and water. All experimental protocols with regard to the use of animals were approved by the Institutional Animal Care Committee of Nanjing First Hospital. All animal experiments were performed in accordance with the guidelines of decreasing the amount of suffering, pain, and discomfort of the experimental animals.
Human clinical samples were obtained from patients who underwent total knee arthroplasties after informed consent and approval from the Ethics Committee of Nanjing First Hospital (Nanjing, China). Only patients with primary knee OA were selected, and patients with inflammatory arthritis or with a history of prior knee surgery were excluded.
Isolation of human ADSCs.
Donor-matched infrapatellar fat pad and subcutaneous fat were obtained from OA patients. Briefly, as for isolation of hADSCs-IPFP and hADSCs-ScAT, the infrapatellar fat pad and subcutaneous fat were harvested and washed in phosphate-buffered saline (PBS), and then finely diced into small pieces by using a surgical scissor. The diced tissues were digested in 0.1% type I collagenase (Sigma, America) for 10 h, and the cell suspension was filtered through a 40-μm cell strainer (BD Bioscience). The released cells were centrifuged at 500 g for 10 min and resuspended in Red Cell Lysis Buffer (Beyotime, China) at room temperature for 10 min. The cells were then centrifuged again, and resuspended in Dulbecco's modified Eagle's medium (DMEM)/F12 (Invitrogen, America) supplemented with 10% fetal bovine serum (FBS; Gibco, America) and 1% P/S. The medium was changed every other day.
Isolation of mouse ADSCs.
8-week-old C57/B6J mice were sacrificed by cervical dislocation under isofurane anaesthesia. Adipose tissue was harvested from the inguinal areas and infrapatellar fat pad used in all experiments. Mouse ADSCs (mADSCs) were isolated from each adipose tissue sample. Briefly, adipose tissue was washed with phosphate-bufered saline (PBS) (pH 7.4) and cut into 0.5–1.0 mm small fragments. The tissues were then transferred into 15-mL tubes. Type I collagenase (1.0 mg/mL was then added to the tube at an identical volume. The mixture was immediately agitated using a water bass shaker (150 rpm) at 37 °C for 30 min. The digested tissue was filtered through a 40-μm cell strainer and centrifuged at 200×g for 5 min. The supernatant was aspirated and the cell pellet was resuspended in erythrocyte lysis buffer (168 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA4Na; 10 mL) at 4 °C for 10 min. After erythrocyte lysis, 5 mL of medium was added, and the tube was centrifuged at 200×g for 5 min. The cell pellet was resuspended in medium and filtered through sterilized 100- and 40-μm cell strainers (Corning Inc., NY, USA). The mADSCs were cultured and used in the third passage.
Identification of ADSCs.
For detecting the classical biomarkers of ADSCs, we performed flow cytometric analysis by the following fluorescein isothiocyanate- (FITC-) conjugated or phycoerythrin-(PE-) conjugated antibodies: CD29, CD44, CD90, CD105, CD73, CD34, and CD45 (Becton Dickinson, San Jose, CA, USA). The FITC-IgG and PE-IgG isotypic immunoglobulins were detected as isotype controls. After incubation, cells were washed twice and finally suspended in FACS buffer for flow cytometry analysis (BD Biosciences).
The multipotent differentiation potential of ADSCs to differentiate to osteoblasts, adipocytes, and chondroblasts was evaluated. After subculturing to the third generation, the culture medium was replaced with osteogenic, adipogenic, or chondrogenic differentiation complete medium (Cyagen Biosciences, China). After the induction of the differentiation cultures for 14 days, the accumulation of calcium, intracellular lipids, and mucopolysaccharides was estimated by the alizarin red staining, oil red O staining, and Alcian blue staining (Sigma-Aldrich, USA), respectively.
Isolation of human-chondrocyte.
Primary human OA chondrocytes were isolated from leftover pieces of osteoarthritic cartilage from total knee replacement surgery of seven OA patients, Articular cartilage was removed aseptically from subchondral bone, cut into pieces, and washed with PBS, then digested overnight in high-glucose DMEM (Invitrogen, America) supplemented with 0.2% type II collagenase (Sigma, America) and 1% P/S. The cell suspension was filtered by a 40-μm cell strainer; and the collected cells were centrifuged at 400 g for 5 min, and then resuspended in high glucose DMEM supplemented with 10% FBS and 1% P/S. The medium was replaced every other day. Chondrocytes were induced with IL-1β (10 ng/ml) for 24 h after transfection for subsequent experiments.
Isolation of mouse-chondrocyte.
To extract chondrocytes, 3-week-old C57/B6J mice were sacrificed for collection of cartilage from knees. First, cartilage was into small pieces after washing with PBS. Second, the samples were digested in 0.25% trypsin-EDTA (Gibco, USA) solution for 5 min, and DMEM-F12 (Gibco) containing 0.2% collagenase type II (Sigma-Aldrich) for 6 h at 37 °C, successively. The released chondrocytes were seeded in T25 cell culture flasks. Cells were passaged at a ratio of 1:3 at 80% confluence. The culture medium was refreshed every 3 days. Chondrocytes were induced with IL-1β (10 ng/ml) for 24 h after transfection for subsequent experiments.
Exosome Extraction and Identification.
The Exos derived from ADSCs were isolated as previously described. In brief, culture media supplemented with exosome-depleted FBS (SBI, USA) were used for cultivating ADSCs. Subsequently, conditioned supernatants from ADSCs cultures were collected and centrifuged such to remove dead cells or debris. The collected medium was subjected to ultracentrifugation at 110,000 g for 2 h at 4℃ after filtration with a 0.22-mm filter. The exosome-containing pellet was washed and resuspended with PBS and stored at -80°C prior to further analysis. The total protein contents of the Exos were evaluated.
Analysis of particle size and intensity was conducted with nanoparticle trafficking analysis (NTA) system (NanoSight NS300). After isolation, the Exos were diluted infiltered PBS before administration. Samples were administered and recorded under controlled flow by the NanoSight syringe pump. Automatic settings were performed to measure the minimum particle size and track length. The measuring conditions were temperature 23.75 ± 0.5°C, 25 frames per second, and measuring time 60s. The detection threshold was uniform in the different groups.
For morphologic observation with transmission electron microscopy (TEM), exosome pellets were seeded on formvar carbon-coated 200-mesh copper electron microscopy grids, placed at room temperature for 5 min, and then were stained with aqueous uranyl acetate. The grids were washed with PBS and continued to semidry at room temperature prior to detection under TEM (Hitachi, H7500 TEM, Tokyo, Japan).
The CD63, CD9, TSG101 and Calnexin biomarkers were measured with western blotting analysis. Exos were collected and resolved by SDS/PAGE and then transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked by 5% nonfat milk in TBST buffer and incubated overnight using rabbit anti-CD63 (1:300, Proteintech, China), CD9 (1:1000, Proteintech, China), TSG101(1:2000, Proteintech, China), Calnexin(1:5000, Proteintech, China) separately, then washed with TBST, and incubated continuously using HRP linked goat anti-rabbit IgG (1:5000, keygen, China), and the protein intensity was determined with the automatic imager (General Electric, Fairfield, CT, USA).
Exosome Label and Track.
To determine whether ADSCs derived-Exos can be taken up by chondrocytes, Exos were labeled with PKH26 (Sigma-Aldrich, USA) following the protocol recommended by the manufacturer. which allow us to fluorescently label isolated Exos to track cellular interaction and uptake. Exos were co-cultured with chondrocytes then fixed with 4% paraformaldehyde. The nuclei were stained with Hoechst 33342 (Beyotime, China). The cytoskeleton was stained by Actin-Tracker Green (Beyotime, China). The uptake of exosome was observed using a confocal laser scanning microscope (Zeiss LSM710, Germany).
Determination of Cell Viability.
Cell viability was quantitatively evaluated by Cell Counting Kit-8 (Beyotime, China). In brief, chondrocytes (1 × 104) were planted in 96-well microculture plates incubated overnight. Then, the viability assay was conducted, and absorbance was assessed using a microplate reader at a wavelength of 450 nm according to the manufacturer’s instructions with some modification. Three independent experiments were performed, and cell viability of different groups was evaluated as a percentage of the control.
Migration assay
Migration in conditioned chondrocytes was detected using a Transwell system. Briefly, 5 × 104 chondrocytes were placed in a 24-well transwell plate with 8 μm pore size inserts (Corning, NY, USA,). Then, 500 μL of DMEM containing 0.5% FBS and 1% PBS with 5 μg Exos was added to the lower chamber before culturing for 16 h. The cells in the upper chamber (2 × 105 cells) were then placed in 4% paraformaldehyde for 20 min and mixed with 0.5% hematoxylin and eosin for 10 min. The percentage of cell migration in each well was determined in a blinded manner.
Cell apoptosis analysis
chondrocytes were plated in 6-well plates at a concentration of 2 × 105/well and were treated with or without ADSC-Exo (5ug). Cell apoptosis was analyzed by using an Annexin V assay kit (Keygen, China) and then measured by a Beamcyte-1026® flow cytometer (BDA Inc., China).
Exosomal small RNA sequence assay
The small RNA sequence for hADSCsScAT Exos and hADSCsIPFP Exos were performed by OE Biotech Company (Shanghai, China). Three samples were processed for each type of exosome. The fragmentation mixtures were hybridized to an Agilent-Human microRNA array 21.0 (8 ×60 K, Design ID:070,156). For microarray analysis, the Affymetrix (Santa Clara, CA, USA) miRNA 4.0 platform was employed. The sample labeling, microarray hybridization and washing were performed based on the manufacturer’s instructions (Agilent Technologies Inc., Santa Clara, California, USA). Differentially expressed miRNAs were identified using a fold change cut off value of ≥1.5 set for both up- and down-regulated genes.
Luciferase Reporter Assay.
Luciferase vectors including the 3′-UTR of ADAMTS4 containing the ADAMTS4-miR-99b-3p response elements and the mutant were obtained from GenePharma. Either miR-99b-3p mimic and negative control was then transfected into the human embryonic kidney (HEK) 293 cells in the presence of either the wild-type or the mutant reporter plasmid. Luciferase activity was determined with the Dual-Luciferase reporter system (Vazyme), and Renilla luciferase activity was set as internal control.
miRNA expression and transfection, miRNA abundant Exos isolation
miR-99b-3p-mimic, miR-mimic-NC, were constructed by RiboBio (Guangzhou, China). The ADSCs and chondrocytes were transfected with mimics at a density of 4 × 105 each well in six-well plates using Lipofectamine 2000 reagent (Invitrogen).
The Exos derived from miR-99b-3p-mimic transfected ADSCs were isolated as previously described.
Plasmid transfection assay
The pcDNA3.1 vector and pcDNA3.1 vector for ADAMTS4 overexpression commercially were prepared by General Biosystems (GenePharma, China). These plasmids were transfected into chondrocytes with the Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Synthesis of HB-PEGDA polymers
Vinyl terminated hyperbranched PEG polymers (HB-PEGDA) were prepared via an in situ reversible addition−fragmentation chain transfer polymerization (RAFT) strategy.[37] Briefly, using 2’-azobis(2-methylpronitrile) (AIBN) as the initiator and tetraethylthiuram disulfide (DS) as the RAFT agent precursor, the polymerisation of polyethylene glycol diacrylate (PEGDA, average Mn = 700) (0.4 mol·L−1) was performed in 75 mL of butanone at 70 °C with a feed ratio of [PEGDA] : [DS] : [AIBN] at 25:1:1.8. Gel permeation chromatography (GPC, Agilent 1260 Infinity II) was used to monitor the evolution of polymer molecular weight during the polymerization. Unless otherwise stated, the following hydrogel syntheses in this work were performed using HB-PEGDA polymer with a molecular weight of 15 kDa. NMR data were collected by a Bruker AVANCE NEO 400 MHz NMR spectrometer.
Synthesis of SH-HA
SH-HA was synthesized using a previously described method.[40] Briefly, HA (500 mg, 1.25 mmol) was dissolved in 10 mL MES buffer (pH 4.75, 0.1 M) with DTP (595 mg, 2.5 mmol) and EDCI (480 mg, 2.5 mmol). The mixture was stirred for 5 h before 1 M NaOH was added to adjust the pH to 7.0 for quenching the reaction. Then, DTT (501 mg, 3.25 mmol) was added, after which the pH of the solution was raised to 8.5 using 1 M NaOH. The mixture was stirred for 24 h. At the end of the reaction, the pH of the solution was adjusted to 3.5 by 1 M HCl. The acidified solution was transferred to dialysis tubes (Mw cutoff = 12 kDa) and dialyzed against dilute HCl (pH 3.5) containing 100 mM NaCl for 4 times. Finally, the solution was centrifuged, and the supernatant was lyophilized to obtain the white fluffy product.
Preparation of HMPs through the microfluidic droplet technique
HMPs were prepared by a Michael addition reaction between HB-PEGDA and SH-HA within discrete droplets produced by a microfluidic droplet maker.[41] Briefly, in a typical experiment, aqueous dispersions containing 1 % (w/v) SH-HA and 3.33 % (w/v) HB-PEGDA were intersected by the fluorinert TM FC-40 oil containing 2 wt% Pico-SurfTM in the microfluidic droplet maker to generate uniform microdroplets. The as-obtained microdroplets were left at room temperature for 1 hour to ensure adequate gelation. Then, to purify the HMPs, 20% v/v 1H, 1H, 2H, 2H-perfluoro-1-octanol (PFO) in Novec 7500 oil was added into the samples to remove the surfactant. The purified HMPs were soaked and washed three times with PBS buffer, and then dispersed in the PBS buffer for later use.
Hydrogel Rheological Characterization
Rheological properties of the samples were assessed using a stress-controlled rheometer (HAAKE RheoStress 1, Thermo Scientific, USA) fitted with an 8 mm diameter flat plate (2 mm gap) at 25℃. Prior to rheological testing, the granular hydrogel prepared by centrifuging HMP dispersion at 104 rpm for 15 min were carefully pipetted into the plate gap. Dynamic oscillatory strain amplitude sweep measurement was conducted by varying % strain from 0.1 to 1000% at a frequency of 1 Hz, and oscillatory frequency sweeps were conducted ranging from 0.1 to 100 rad/s at 1% strain amplitude.
Exos encapsulation efficiency
The amount of remaining Exos in the solution was detected by BCA protein assay. The following formulas were used for calculating the Exos encapsulation efficacy (EE):

Cell cytotoxicity and compatibility
Chondrocytes were seeded in a 96-well plate at a density of 1×104 cells/well and cultured overnight, HMPs, HMPs@ExosScAT, HMPs@ExosScAT-99b-3p were added to each well. After 24 h, cell viability was conducted using the Cell Counting Kit-8 (CCK-8, Beyotime, China) assay.
In addition, the cyto-compatibility was assessed by Calcein AM/PI staining assay. chondrocytes at passage three were seeded in 24-well plates at a density of 5×104 cells/well and cultured overnight prior to the addition of 1-5 mg/mL. After 1, 2, and 3 days, the viability of the cells was analyzed using the Calcein/PI Cell Viability/Cytotoxicity Assay Kit (Beyotime, China).
Immunofluorescent Staining.
For cell immunofluorescence, chondrocytes were incubated in complete culture medium with various Exos for an additional three hours. The cells were then washed in phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 15 min. The fixed cells were permeabilised with 0.1% Triton X-100 for 5 min and washed with PBS three times. Then, the cells were treated with primary ADAMTS4, ACAN, COMP antibodies (Abcam, Cambridge, UK) overnight at 4°C in PBS supplemented with 1% bovine serum albumin.
Immunohistochemistry staining
Immunohistochemical analysis was performed as described previously [31]. Briefly, samples were fixed in 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA), decalcified, embedded in paraffin, and cut into 5-μm sections that were deparaffinized, rehydrated, and then incubated with primary ADAMTS4, ACAN, COMP antibody (Abcam, Cambridge, UK) overnight at 4 °C. Sections were incubated with secondary antibody at 37 °C for 1 h and with 3,3-diaminobenzidine (DAB) for 3 min.
Evaluation of an in Vivo Imaging System (IVIS)
To determine the residence time of Exos, ExosScAT-99b-3p, HMPs@Exos and HMPs@ExosScAT-99b-3p in the articular cavity. Exos were labelled with PKH26. Exos (50μg), ExosScAT-99b-3p (50ug), HMPs@exo (50ug Exos/), HMPs@ExosScAT-99b-3p were injected into right knee of mice, respectively. Then, the mice were scanned by the IVIS Lumina II system (PerkinElmer, USA) to observe the retention time of Exos in all groups.
Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR).
After harvesting the chondrocytes and separating the total RNA by TRIzol Reagent (Invitrogen), and reverse transcribed into complementary DNA according to the manufacturer’s protocol of the reverse transcription kit (R312, Vazyme, China). qPCR was carried out following the instructions provided by the Taq Pro Universal SYBR qPCR Master Mix (Q712, Vazyme, China) using a real-time fluorescence quantitative PCR appliance (QuantStudio®5, ThemoFisher Scientific, USA). Otherwise, the expression of miRNAs was quantified by miRNA Universal SYBR qPCR Master Mix (Vazyme, China) and miRNA 1st Strand cDNA Synthesis Kit (Vazyme, China) was applied for the single stranded cDNA synthesis. GAPDH and U6 were taken as internal reference genes. All the primers for RNAs are listed in Additional file: Table 1.
Table 1 Primer sequence for RT-qPCR
|
Genes |
Primer sequence |
|
miR-381-3p |
F:CGGGCTATACAAGGGCAAGC R:CAGCCACAAAAGAGCACAAT |
|
miR-224-5p |
F:CGGGCTCAAGTCACTAGTGGTTC R:CAGCCACAAAAGAGCACAAT |
|
miR-99b-3p |
F:CGGGCCAAGCTCGTGTCTGT R:CAGCCACAAAAGAGCACAAT |
|
miR-27a-5p |
F:CGGGCAGGGCTTAGCTGCTT R:CAGCCACAAAAGAGCACAAT |
|
miR-889-3p |
F:CGGGCTTAATATCGGACAA R:CAGCCACAAAAGAGCACAAT |
|
miR-424-3p |
F:CGGGCCAAAACGTGAGGCG R:CAGCCACAAAAGAGCACAAT |
|
miR-744-5p |
F:CGGGCTGCGGGGCTAGGGCT R:CAGCCACAAAAGAGCACAAT |
|
miR-483-5p |
F:CGGGCAAGACGGGAGGAAAG R:CAGCCACAAAAGAGCACAAT |
|
miR-222-3p |
F:CGGGCAGCTACATCTGGCT R:CAGCCACAAAAGAGCACAAT |
|
miR-503-5p |
F:CGGGCTAGCAGCGGGAACAGT R:CAGCCACAAAAGAGCACAAT |
|
miR-29a-5p |
F:CGGGCACTGATTTCTTTTGG R:CAGCCACAAAAGAGCACAAT |
|
U6 |
F: 5′-GGTCGGGCAGGAAAGAGGGC-3′ R: 5′-GCTAATCTTCTCTGTATCGTTCC-3′ |
Western Blot Analysis
The proteins were harvested and lysed in RIPA buffer. Equal amounts of protein extracts (30 μg) were loaded per lane and resolved by SDS/PAGE. Subsequently, polypeptides were separated and transferred to PVDF membranes. The membranes were blocked and then treated overnight with rabbit anti-Col2a1 (1:1000, Abcam, UK), rabbit anti-MMP13 (1:1000, Cell Signaling Technology), rabbit anti-ACAN (1:1000, Abcam, UK), rabbit anti-COMP (1:1000, Abcam, UK), rabbit anti-ADAMTS4 (1:1000, Abcam, UK), and mouse anti-GAPDH (1:1000, Beyotime) antibodies, respectively. After being washed with TBST three times, the membranes were incubated using the corresponding HRP-conjugated secondary antibodies (1:1000, Beyotime) with blocking solution at room temperature for 2 h. Finally, the expression levels of protein were measured by enhanced chemiluminescence kit (Millipore). The protein bands were normalized by GAPDH and quantified as the ratio of the optical density.
Induction of OA
All animal experiments were in accordance to the Animal Research Committee regulations of Nanjing first hospital, Nanjing medical university. OA was induced in 8-week-old male C57BL/6 mice by surgical destabilization of the medial meniscus (DMM) of the right knee. Specifically, the DMM surgery was performed by surgical sectioning of the medial meniscotibial ligament and the sham operation was performed by incision of the cutaneous and muscular planes at baseline.
Intra‐articular injection
In the animal section 1 in Fig. 2, all mice underwent DMM surgery or sham surgery were randomly divided into three groups: (1) PBS group (control); (2) ExosScAT group; (3) ExosIPFP group; (4) ExosScAT-NC group; (5) ExosScAT-99b-3p group. The mice were given intra-articular injections of 10μl PBS or 10μl Exos (5ug) twice a week for 4 weeks with a 30‐gauge needle and a micro syringe (Hamilton). Afterward the mice were sacrificed, and cartilage tissue was dissected for histological assessment (H&E, Safranin O/fast green and toluidine blue staining) and immunofluorescent staining (ADAMTS4, ACAN, COMP, Col2a1).
In the animal section 2 in Fig. 6, all mice underwent DMM surgery were randomly divided into five groups: (1) PBS group (Control); (2) ExosScAT-99b-3p group; (3) HMPs group (4) HMPs@ExosScAT group; (5) HMPs@ExosScAT-99b-3p group. The mice were injected with 20μl PBS, 20μl Exo@ScAT-99b-3p (20ug), 30μl HMPs, 30μl HMPs@Exos (containing 20ug Exos), 30μl HMPs@ExosScAT-99b-3p (containing 20ug Exos) at the first day. After 4 weeks, the mice were sacrificed, and cartilage tissue was dissected for histological assessment (H&E, Safranin O/fast green and toluidine blue staining) and immunofluorescent staining (ADAMTS4, ACAN, COMP, Col2a1).
Isolation and identification of hADSCsScAT and hADSCsIPFP.
We obtained human IPFP tissues and ScAT tissues from OA donors with total knee arthroplasty and isolated MSCs according to the methods described in previous study. Then, the isolated ADSCs were observed under an optical microscope, and the image in Fig. 1A shows that the two kinds of ADSCs displayed typical MSC morphology with a spindle-like shape. Moreover, flow cytometry analysis showed that more than 95% of these cells expressed classical MSC markers, including CD73, CD90, and CD105, while no cells expressed haematopoietic markers, such as CD34, CD45, and HLA-DR (Fig. 1B), indicating a high enrichment of ADSCs. Next, we used an in vitro differentiation assay to further investigate whether these cells have the potential to differentiate into adipocytes, osteocytes, and chondrocytes. Osteogenic potential was confirmed by the staining of calcium mineral deposits with Alizarin Red (Fig. 1A), while adipogenic potential was evaluated by the observation of small cytoplasmic lipid droplets stained with Oil Red O (Fig. 1A). Chondrogenic potential was confirmed by staining with sulfated glycosaminoglycans using Alcian Blue (Fig. 1A).
Isolation and identification of Exos derived from hADSCsScAT and hADSCsIPFP
Transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA) and western blotting were used to characterize the Exos secreted from ADSCs. TEM clearly revealed that Exos exhibited a cup-shaped or round morphology with a diameter of 50–150 nm (Fig. 1C). NTA showed that most of these vesicles ranged from 30 to 150 nm in size (Fig. 1C). Western blotting analyses indicated that ExoScAT and ExoIPFP expressed exosomal markers such as CD9, CD63, and TSG101 (Fig. 1D).
Uptake of ADSC-Exos by chondrocytes
The laser confocal microscopy image showed the accumulation of Exos in chondrocytes. The nuclei of chondrocytes were stained with DAPI (blue), actin of chondrocytes was stained with phalloidin-FITC (green), and the Exos were labelled with PKH26 (red). The Exos were observed in the perinuclear region of the chondrocytes (Fig. 1E), confirming the successful internalization of ADSC-Exos by chondrocytes. Moreover, there was no difference between ExosScAT and ExosIPFP internalized by chondrocytes according to the fluorescence intensity.
ADSC-Exos promote proliferation, migration, and anabolism and inhibit apoptosis and catabolism in osteoarthritic chondrocytes
To investigate whether ADSC-exos alleviate OA by targeting chondrocytes, we harvested ExosScAT and ExosIPFP; we added different Exos at concentrations of 50 μg/ml and 100 μg/ml to IL-1β (10 ng/ml)-treated chondrocytes cultures for 7 days. The results from CCK-8 assays showed that the proliferation of chondrocytes was stimulated by Exos in a dose-dependent manner (Fig. 2A); however, there was no significant difference between ExosScAT and ExosIPFP at the same concentration (Fig. 2A). Transwell assays and Annexin V-FITC/PI staining were performed to evaluate migration capacity and apoptosis in IL-1β (10 ng/ml)-treated chondrocytes. The results showed that Exos promoted chondrocyte migration and inhibited chondrocyte apoptosis; however, no significant difference between ExosScAT and ExosIPFP was observed (Fig.2B~E). Furthermore, we measured the protein levels of anabolism-related genes (collagen II and aggrecan) and catabolism-related genes (MMP13 and ADAMTS4), which are highly related to OA pathological processes [51]. IL-1β treatment significantly decreased collagen II and aggrecan expression, while it increased the protein levels of MMP13 and ADAMTS4. The coculture of Exos with chondrocytes reversed the effect of IL-1β on aggrecan and ADAMTS4. Interestingly, the expression of aggrecan in the ExoIPFP group was higher than that in the ExoScAT group, while Col2a1 and MMP13 levels were similar in the two groups (Fig. 2 F, G).
MiR-99b-3p is upregulated in ExoIPFP and plays a critical role in maintaining anabolism in articular chondrocytes.
To further investigate the mechanism by which Exos affect chondrocytes, we isolated RNA from ExosIPFP and ExosScAT, performed small RNA sequencing of the miRNAs derived from the Exos and compared them between the two groups (Fig. 3A). According to the sequencing results, we identified the 10 miRNAs that had the most obvious upregulation and further verified them in the Exo fractions by RT‒qPCR. These results showed that miR-99b-3p was the most upregulated miRNA in ExosIPFP.
MiR‑99b‑3p regulates ADAMTS4 by directly targeting the 3′‑UTR.
To identify the target gene for miR-99b-3p, the potential regulatory relationship between mRNA and miRNAs was predicted using the TargetScan, miRWalk, miRTarBase, and miRPathDB databases. Because the changes in chondrocytes mainly occur in aggrecan, one of the components of the ECM, we searched the gene set related to ECM expression in the GO database (GO:0031012). Then, we calculated Venn diagrams (Fig. 3C) to identify the overlap between different genes and miRNA data in the database, and we predicted that ADAMTS4 was a potential target of miR-99b-3p. As shown in Fig. 3D, miR-99b-3p has one potential complementary sequence with ADAMTS4. A luciferase reporter assay was used to confirm the direct target of miR-99b-3p, as shown in Fig. 3E. The luciferase activity of ADAMTS4-Wt was suppressed when the cells were cotransfected with the miR-99b-3p mimic, while this effect was not observed for ADAMTS4-Mut, suggesting that ADAMTS4 is a direct target of miR-99b-3p.
The expression of miR‐99b‐3p was markedly reduced in OA cartilage compared to normal cartilage (Fig. 3F). However, higher protein levels of ADAMTS4 were observed in OA cartilage than in normal cartilage (Fig. 3G, H) Moreover, in IL-1β-induced chondrocytes, miR‐99b-3p expression was decreased, and ADAMTS4 protein levels were increased (Fig 3I~K).
Histological staining of cartilage in OA patients and normal patients showed that the cartilage surface of OA patients was severely worn, and safranin O/fast green staining indicated a great loss of glycosaminoglycan in OA cartilage (Fig. 3L). Through immunohistochemical staining, we found that the expression of ADAMTS4 in OA cartilage was significantly higher than that in normal cartilage, while the expression of ACAN and COMP was significantly lower in OA samples (Fig. 3L).
MiR‑99b-3p promotes cartilage-related protein expression by targeting ADAMTS4 in vitro
Chondrocytes were transfected with miR-99b-3p mimic and miR-NC, miR-99b-3p expression was assessed by RT‒qPCR, and we discovered that the miR-99b-3p mimic elevated miR-99b-3p expression nearly 8 times compared with the NC group (Fig. 4A). ADAMTS4 expression was quantified using western blot analysis (Fig. 4B, C). The results showed that ADAMTS4 expression was reduced by the miR-99b-3p mimic. To further explore the relationship between miR-99b-3p and ADAMTS4, in vitro rescue experiments were performed. We transfected the miR-99b-3p mimic or miR-NC into chondrocytes and then cotransfected the chondrocytes with a plasmid that overexpresses ADAMTS4 (pcDNA-ADAMTS4) or with pcDNA-NC. The results showed that upregulating miR-99b-3p restored the protein levels of ACAN and COMP inhibited by pcDNA-ADAMTS4. Together, these results demonstrated that miR-99b-3p targets and negatively regulates ADAMTS4 (Fig. 4D~G), while miR-99b-3p overexpression exhibited a positive effect on the expression of cartilage-related proteins that were inhibited by ADAMTS4 (Fig. 4D~G).
Overexpression of miR-99b-3p increases anabolism-related protein expression more potently in ExosScAT than in in ExosIPFP in vitro
Next, the effect of ADSC-derived Exos on chondrocytes was investigated. ADSCsScAT were transfected with miR-99b-3p mimic and miR-NC, and miR-99b-3p expression was assessed by RT‒qPCR. The results showed that miR-99b-3p expression in the ADSCsIPFP group was approximately 3 times higher than that in the ADSCsScAT group, while the ADSCsScAT-miR-99b-3p group was approximately 12 times higher than that in the ADSCsScAT group (Fig. 4H). Then, we isolated Exos from the corresponding ADSCs and assessed miR-99b-3p in all groups. Compared with the ExosScAT and ExosIPFP groups, miR-99b-3p expression increased more than 11-fold and 4-fold in the ExosScAT-99b-3p group, respectively (Fig. 4I). Chondrocytes were stimulated with IL-1β for 24 h, followed by treatment with different kinds of Exos (ExosScAT, ExosIPFP, ExosScAT-NC, ExosScAT-miR-99b-3p) (Fig. 4J). The results showed that the expression of ADAMTS4 was lower in all exosome treatment groups than in the control group. The ExosIPFP group was lower than the ExosScAT group, while the ExosScAT-99b-3p group had the lowest expression. Additionally, the expression of ACAN and COMP was higher in all treatment groups than in the control group, and the expression in the ExosScAT-99b-3p group was significantly higher than that in the ExosScAT and ExosIPFP groups (Fig. 4K~N).
Overexpression of miR-99b-3p in ExosScAT promoted cartilage regeneration in mouse DMM models
We evaluated the effects of intra-articular injected exosome treatments in mice with OA by histological assessment (Fig. 5A). Four weeks after the DMM operation, the knee joint specimens of animals were collected for H&E, Safranin O/Fast Green staining and Toluidine blue staining (Fig. 5B). In the control group, the cartilage surface was rough and badly worn, the articular cartilage was thin or even missing, and the subchondral bone was exposed and destroyed. Moreover, significant loss or thinning of the proteoglycans was observed in the control group. The cartilage surface in the ExosScAT and ExosIPFP groups was relatively smooth compared to that in the control group, which had no exposed subchondral bone and increased proteoglycan content; the proteoglycan distribution of the ExosIPFP group was more uniform and denser. However, the ExosScAT-99b-3p group showed the best repair effect, the cartilage surface was smooth, the cartilage thickness was uniform, there were no cracks on the surface; the subchondral bone was well reconstructed, the cells in the repaired tissue were clearly arranged in a vertical column, and the proteoglycan content was significantly higher than that in the other groups, with a uniform distribution. Histological scores, OARSI grade, and Mankin’s scores (Fig. 8C–E) in DMM knees were assessed to quantify the extent of cartilage injury, and the data showed that ExosScAT-99b-3p exhibited a more evident protective influence on cartilage repair after DMM surgery (Fig. 5C~E).
The levels of ADAMTS4, ACAN, COMP, and Col2a1 in the repaired tissue were verified by immunofluorescence staining. With the increased expression of miR-99b-3p in chondrocytes, the expression of ADAMTS4 was decreased, while the expression of ACAN and COMP was increased (Fig. 5F) In other words, the expression of ADAMTS4 was lowest; moreover, ACAN and COMP levels were significantly higher in the ExosScAT-99b-3p group than in the other groups (Fig. 5F). The results showed that overexpression of miR-99b-3p in Exos can attenuate OA by downregulating ADAMTS4 protein in vivo.
Characterization of HMPs
Vinyl-terminated hyperbranched PEG polymers (HB-PEGDA) were prepared via an in situ RAFT polymerization strategy using 2’-azobis(2-methylpronitrile) (AIBN) as the initiator and tetraethylthiuram disulfide (DS) as the RAFT agent precursor (Fig. 6A). The characteristics of the as-obtained products collected at different time points are shown in Figure 6B and 6C. A unimodal weak peak with Đ (Mw/Mn) <1.5 was detected after 1 h of reaction, and then the molecular weight of the polymer steadily increased with time. As the reaction proceeded at 70℃ over 5 h, the molecular weight of the HB-PEGDA polymer increased dramatically with time, and a broad Đ was observed, indicating the formation of HB-PEGDA polymers with hyperbranched structures (Fig. 6D). The PBA-HB-PEGDA polymer with a molecular weight of 15 kDa was used for the following HMP synthesis.
Rheological studies revealed that the storage modulus values (G’) for all three formulations were higher than the loss modulus values (G’’). Moreover, all the samples showed a linear viscoelastic region at low strain percentages with G’ independent of the applied deformation (Fig. 6E). These characteristics indicate that the as-obtained granular hydrogels behaved like viscoelastic gels. Additionally, by controlling the HB-PEGDA:SH-HA ratio, the mechanical properties of the granular hydrogels composed of HMPs can be readily tuned with G’ values varying from 60 to 400 Pa, facilitating the mimicking of mechanical variability in a range of soft tissues (Fig. 6F). As shown in Fig. 6G, HMPs and HMPs@Exos were prepared by microfluidic technology, and gelation formed spontaneously through the thiol-ene Michael addition reaction (Fig. 6H).
The obtained HMPs and HMPs@Exos were observed under a bright-field microscope (Fig. 7A), with the majority of particles being approximately 120 μm in diameter (Fig. 7B).
Exosome loading, distribution, release, and HMP degradation
After labelling PEGDA with fluorescein O-methacrylate and labelling Exos with PKH26, HMPs were prepared. Confocal microscopy showed that the microspheres were uniformly distributed green, and the Exos were uniformly distributed in red dots (Fig.7D). The results showed that PEGDA and Exos were intact and uniformly distributed in the microparticles. Hyaluronidase can act on β-N-acetylhexosamine-1,4 glycosidic bonds, leading to the degradation of microparticles [54–56]. The encapsulation efficiency of Exos in HMPs was 78.5±2.4%.
To investigate the degradation of HMPs in vitro, a degradation assay was carried out in PBS containing hyaluronidase (100 U/mL). During long incubation times, the HMPs containing hyaluronidase showed continuous and slow biodegradation. On the first day, the morphology of the HMPs was almost unchanged. On the 7th day, although most of the HMPs were still intact, a few HMPs started to break. On the 14th day, most HMPs began to collapse, and a large number of gel fragments appeared. On the 28th day, most of the HMPs disappeared completely. However, the HMPs without hyaluronidase remained intact until 28 days (Fig. 7C).
Biocompatibility of HMPs in chondrocytes, sustained release of Exos from HMPs and exosome uptake by chondrocytes
In the no contact coculture system (Fig. 8A), Exos, HMPs, and HMPs@Exos were added to the upper transwell chambers. The laser confocal microscope image (Fig. 8B) showed exosome uptake by chondrocytes. The amount of Exos taken up by chondrocytes in the Exo group was higher on day 1, and then the exosome uptake gradually decreased. At day 7, it was already difficult to detect exosome fluorescence in chondrocytes, whereas in the HMPs@Exos group, although the amount of exosome uptake by chondrocytes was relatively low on day 1, the fluorescence gradually increased in the following days, which was related to the gradual degradation of the hydrogel and the controlled release of Exos. Calcein AM/PI staining of chondrocytes was visualized under a fluorescence microscope; living cells showed green fluorescence, while dead cells showed red fluorescence. The results showed that chondrocytes exhibited good cell viability during 7 days of culture with a negligible number of dead cells (Fig. 8C). CCK-8 assay was used to assess the cytotoxicity of the HMPs in chondrocytes, revealing that the HMPs did not exhibit any significant cytotoxicity when used to treat chondrocytes, with survival rates remaining >90% (Fig. 8D).
Additionally, in the contact coculture system (Fig. 8E), HMPs were cocultured directly with chondrocytes in petri dishes. The LSM images (Fig. 8F) showed that chondrocytes grew on the surface of the HMPs, and the cells of all groups gradually increased over time, while the number of cells in the groups with HMPs encapsulated with Exos was obviously higher than that in the HMPs group. All groups showed a green live cell fluorescence signal with few dead cells. After incubation for 7 days, DAPI/FITC-phalloidin fluorescence staining indicated that chondrocytes covered the HMPs surfaces with exosome uptake into cells (Fig. 8G).
Intraarticular retention and in vivo biological safety of HMP treatment
To determine the intraarticular retention of Exos with or without encapsulation in HMPs, the fluorescent signals were monitored using the IVIS system at 1, 3, 7, 14, and 28 days after intraarticular injection. The signals in both groups decreased over time; however, fluorescence was found in the HMPs@Exos and HMPs@ExosScAT-99b-3p groups on the 28th day, while the signal in the exosome group was undetectable after the 7th day of injection (Fig. S1). These results demonstrated the instability and poor local retention ability of the Exos, but encapsulation in HMPs might improve these shortcomings.
To evaluate the biological safety of the HMPs in vivo, the main organs (heart, liver, spleen, lung, and kidney) were obtained from mice on day 28 after treatment and stained with haematoxylin and eosin (H&E). Comparisons with the control group showed no significant tissue damage or any inflammatory lesions in H&E staining of major organs from the groups (Fig. S2), suggesting that HMPs show good biocompatibility in vivo.
Expression of ADAMTS4 and its substrates (ACAN and COMP) in vitro
Next, the coculture of HMPs and chondrocytes was performed using Transwell plates to examine the effects of different HMPs groups (Fig.). IL-1β-induced chondrocytes were treated with PBS, ExosScAT-99b-3p, HMPs, HMPs@ExosScAT and HMPs@ExosScAT-99b-3p. On the 14th day, the protein expression of ADAMTS4, ACAN, and COMP was determined by immunofluorescence staining and western blotting. The expression of ADAMTS4, ACAN, and COMP in chondrocytes was detected by immunofluorescence (Fig. 9A). The results showed that the HMPs@ExosScAT-99b-3p group expressed less ADAMTS4, while ADAMTS4 was significantly increased in the control and HMPs groups. However, the expression of ACAN and COMP was significantly enhanced in the HMPs@ExosScAT-99b-3p group compared to that in the remaining groups, indicating that HMPs@ExosScAT-99b-3p could effectively inhibit ADAMTS4 expression and promote ECM synthesis in chondrocytes by releasing miR-99b-3p. Consistent with the IF results, the WB results showed that HMPs@ExosScAT-99b-3p significantly inhibited ADAMTS4 protein expression and increased ACAN, COMP and collagen II protein levels compared with those in the other groups on the 14th day of coculture (Fig. 9B, C)
In Vivo Therapeutic Effect of OA
To further investigate the protective effect of HMP-encapsulated Exos on cartilage degeneration, histological and immunofluorescence evaluations were performed at 4 weeks after surgery (Fig. 10A). The articular cartilage in the control group (PBS) showed severe surface abrasion, disorganized chondrocytes, and weak toluidine blue and Safranin O/fast green staining. In comparison, the ExosScAT-99b-3p group and HMPs@ExosScAT group had relative integrity and regular cartilage structure compared with the PBS group, while the HMPs@ExosScAT-99b-3p group showed the best morphological integrity and reduced erosion of cartilage surface destruction. According to the results of Safranin O/Fast Green and toluidine blue staining in the treatment groups, the amount of glycosaminoglycan was highest in the HMPs@ExosScAT-99b-3p group, followed by the HMPs@ExosScAT and ExosScAT-99b-3p groups (Fig. 10B, C). The evaluation of histological scores, OARSI grade, and Mankin’s scores showed that the HMPs@ExosScAT-99b-3p group had the best outcome for maintaining the cartilage ECM (Fig. 10F~G).
Finally, the protein expression levels of ADAMTS4, ACAN, COMP, and Col2a1 were measured (Fig. 10 D, E). The results showed that ACAN, COMP, and Col2a1 protein expression was significantly decreased in the control group compared to the ExosScAT-99b-3p, HMPs@ExosScAT, and HMPs@ExosScAT-99b-3p groups. Compared to that in the control group, the expression of ADAMTS4 in all the groups was decreased. However, the expression of ADAMTS4 treated with HMPs@ExosScAT-99b-3p was the lowest, and the levels of ACAN, COMP, and Col2a1 were highest among all the OA models.
OA is a prevalent joint disease of inflammation that often occurs in the elderly population. According to data from the United States Centers for Disease Control and Prevention, OA affected 52.5 million people in the US in 2012 and is expected to affect 78 million people by 2040.[42] To date, the treatment for OA involves pain management using nonsteroidal anti-inflammatory drugs (NSAIDs) and pain killers to relieve the symptoms and surgical therapy for end-stage OA patients.[9, 43] However, surgical therapy is costly and may lead to tissue hypertrophy. Therefore, appropriate therapeutic strategies are crucial to overcome existing problems.
Homeostasis of the articular cartilage is maintained via a delicate balance among a series of processes.[44] Chondrocytes are the only cell type that forms articular cartilage and maintains a dynamic equilibrium between the synthesis and degradation of ECM components, including type II collagen (Col-II) and aggrecan (ACAN), under normal conditions. However, in an osteoarthritic state, chondrocytes are activated by various cytokines to express an abnormal catabolic phenotype, resulting in accelerated degradation and limited synthesis of the ECM.[20] An increasing number of studies have shown that proteinases play an important role in the development of OA.[45, 46] ADAMTS4, termed aggrecanase-1, is an important cartilage matrix–degrading enzyme that can degrade ECM components.[46, 47] Aggrecanases are the main proteinases present in growth plate cartilage and articular cartilage and are involved in the degradation of proteoglycans. Aggrecanase activity is thought to be responsible for the degradation of cartilage in inflammatory joint diseases. Aggrecanase is considered a marker of cartilage degeneration in inflammatory joint diseases such as OA.[45] ACAN and COMP are the main components of cartilage ECM, which provide cartilage with resistance to compressive stress.[46] Moreover, ACAN and COMP are substrates of ADAMTS4. Aggrecanase-mediated ACAN production is a significant event that occurs in the early stage of OA, following irreversible ECM degradation. Through the synthesis and breakdown of ECM macromolecules, chondrocytes maintain a dynamic balance between cartilage structure and the cellular environment. Once the degradation rate of ECM macromolecules exceeds the synthesis rate, the amount of cartilage matrix decreases, eventually leading to total or partial erosion of cartilage.[48]
Degradation of ACAN is an important marker of OA.[49] Aggrecanase inhibitors have been identified as candidates for arthritis therapy in vivo.[45, 46] Preventing ACAN loss by inhibiting aggrecanase activity can slow progressive cartilage erosion, which may delay or even prevent the development of end-stage OA.[46] Therefore, agents targeting ADAMTS4 may promote ECM homeostasis. Low accumulation of ACAN inside the ECM has been revealed to be a key issue in damaged cartilage tissue. As far as the importance of ECM, miR-99b-3p may be a tool that can be used to maintain its stability by inhibiting ADAMTS4 expression. The expression levels of ACAN and COMP were notably elevated after miR-99b-3p overexpression.
By using an IL-1β-induced in vitro OA model, we demonstrated that ExosScAT−99b−3p have significantly greater efficacy in alleviating chondrocyte degradation than ExosIPFP and ExosScAT, and the chondroprotective effect of ExosScAT−99b−3p may in part be attributed to the downregulation of ADAMTS4. An in vivo experiment using a DMM OA model confirmed that IA injection of ExosScAT−99b−3p resulted in significant alleviation of OA progression in cartilage. Our study may therefore introduce a new approach for the treatment of OA.
Many reports have shown that miRNAs carried by Exos secreted by stem cells can alleviate the progression of OA.[18, 22, 23] In clinical therapy, ADSCs are superior to other cells because of their lower immune rejection potential, abundant sources, safety and low cost.[8, 9] In this study, we found that there were some differences in ECM synthesis and degradation between IPFP- and ScAT-derived Exos in the treatment of OA. After sequencing miRNA in the two kinds of Exos, we found that miR-99b-3p partially account for the difference, so we further studied miR-99b-3p by overexpressing miR-99b-3p in ExosScAT, which significantly promoted the expression of ECM-related protein (ACAN, COMP) in OA chondrocytes in vitro and DMM models in vivo.
Naked miRNA is susceptible to degradation by RNase in the extracellular environment.[50] Therefore, we used stem cell-derived Exos to deliver miR-99b-3p. Exos secreted by MSCs have a large number of microRNAs (miRs), which can be phagocytosed by chondrocytes to regulate cell function.[18, 22, 25] Hence, new cell-free therapies based on exosomal miRs secreted by stem cells are considered a safe and effective alternative to stem cell therapies for OA. However, when Exos are injected in vivo, they can be washed away easily due to the lack of a supporting matrix, which is the major challenge for using Exos in clinical applications.[50, 51]
Despite these promising new approaches to enhance exosome efficacy, the short half-life of these nanoparticles following systemic administration hinders their therapeutic utility.[52] Additionally, local administration of Exos into the defect has only transient results, and successive injections are often required for clinical benefit.[53] The use of injectable biomaterials to facilitate the delivery of Exos and improve their bioavailability in situ has attracted growing interest.[54] Several studies have reported the controlled release of Exos from biomaterial systems.[55, 56] Ideally, these systems should be deliverable in a minimally invasive manner and enhance vesicle retention in situ to facilitate exosome-induced cartilage formation.
Recently, a novel hydrogel formulation with good biocompatibility and injectability was developed based on the spontaneous Michael addition reaction between vinyl-terminated PEG and thiolated polymers, serving as diverse tissue scaffolds.[37, 38] Moreover, in combination with the microfluidic technique, HMPs were also prepared from the microdroplet templates, and granular hydrogels were assembled through packing of the as-obtained HMPs using either centrifugation or vacuum-driven removal of the liquid phase.[45] Accordingly, HMPs composed of HB-PEGDA and SH-HA were obtained in this work for OA treatment through intra-articular injection. HMPs have been widely used in biomedical research for cartilage repair, drug delivery and cell-based therapy. Yu et al. combined a microfluidic method with a photopolymerization process and developed cartilage-targeting peptides and ROS-responsive nanoparticles, which showed excellent potential to repair OA.[38] Han et al. prepared GelMA hydrogel microspheres encapsulated with the anti-inflammatory drug diclofenac sodium (DS) via microfluidic technology to achieve sustained local drug release to treat OA.[57] HMPs have a number of unique properties compared to bulk hydrogels that make them attractive for biomedical applications, and they have emerged as a novel and efficient drug delivery system for injection therapy.[58] First, their small size enables injection through small needles and catheters and inhalation of particles, which is advantageous for minimally invasive delivery of cells and biologics.[59] Second, HMPs can encapsulate different molecules or drugs and gradually release them by controlling their own degradation rate.[57] Third, properly sized HMPs provide good drug protection by minimizing clearance by blood vessels or lymphatics.[58] Therefore, we developed a novel approach to enhance the action time of Exos and ExosScAT−miR−99b−3p by encapsulation in a hydrogel microparticle system.
Repeated and regular injections tend to increase the incidence of adverse events, such as pain, joint bleeding and infection.[60] HMPs have the characteristics of biocompatibility, mechanical stability and minimal invasion (via syringe needles).[38, 57, 58] Yang et al reported that hydrogel microspheres can stay in the articular cavity for a long time, which can effectively eliminate the need for repeated injection.[61] The degradation properties of intra-articular injected biomaterials are important for OA treatment. Good biodegradation activity is a necessary requirement for intra-articular drug carriers. We investigated the degradability and exosome release kinetics of HMPs to evaluate their use as a drug carrier delivery system for long-term release. Hyaluronidase can act on the HA component in HMPs, leading to degradation of the HMPs and simultaneous release of the Exos. We studied the degradation of HMPs in vitro using PBS containing 100 U/mL hyaluronidase, and the degradation and absorption properties were studied in vivo by intra-articular injection. Both in vivo and in vitro studies showed a degradation rate of the HMPs approaching nearly 28 days and predicted a slow release of Exos up to 28 days, which is very beneficial for long-term cartilage repair.
There are multiple pendant vinyl groups in hyperbranched polymers (HBPs) that can be used for functionalization through a variety of chemistries, such as photochemistry and thiol − ene Michael addition. A hyperbranched poly (poly(ethylene glycol) diacrylate) (HB-PEGDA) was synthesized by in situ RAFT polymerization using DS as the RAFT agent precursor, and HB-PEGDA can easily cross-link with SH-HA. HB-PEGDA and SH-HA are nonimmunogenic and have good biocompatibility. In addition, further functional groups can be introduced into the hydrogel to enhance its functionality for specific applications.[37, 62] At present, there are several methods to prepare HMPs, including emulsions, microfluidics, and electrospray methods. The microfluidic technique has obvious advantages in forming HMPs with controllable particle sizes and monodisperse particle sizes by adjusting the channel geometry and input flow rate. Additionally, the characteristics of HMPs can be used to encapsulate various functional components to play a specific role. In this study, we used a microfluidic technique to prepare HMPs with a uniform size and diameter of approximately 100 µm, which allowed the HMPs to smoothly pass through the syringe needle without resistance during injection. Our animal experiments confirmed that the HMPs prepared by microfluidic technology have good injectability, and the encapsulated Exos are well protected, facilitating maximal efficacy.
In the present study, we reported that ExosIPFP appeared to be more effective than ExosScAT in treating some aspects of OA. Our small RNA sequencing results suggest that miR-99b-3p may play a role in improving the integrity of the ECM. ADAMTS4 was predicted as a possible target gene of miR-99b-3p via several databases. When ADAMTS4 expression was inhibited, abundant miR-99b-3p in Exos enhanced the expression of the corresponding components in the ECM and improved the therapeutic effect in OA. In future clinical applications, the overexpression of miR-99b-3p in ExosScAT will enable them to be as effective as or more effective than ExosIPFP in the treatment of OA, and the abundance of subcutaneous adipose tissue makes it possible to extract a large number of stem cells and corresponding Exos.
Moreover, to solve the problem of the short half-life of Exos in vivo, we used microfluidic technology to prepare HMPs to encapsulate Exos. The HMPs had a favourable ability to extend exosome retention and biocompatibility owing to the hydrogel and could effectively promote ECM synthesis in chondrocytes. In mouse OA models, HMPs were effectively degraded, and miR-99b-3p-abundant Exos were gradually released, inhibiting the degradation of the ECM and promoting the repair of damaged cartilage in OA. Together, the miRNA-abundant exosome delivery HMPs system, developed via the microfluidic method, has great potential as a novel strategy for treating OA.
Ethics approval and consent to participate
All experimental protocols with regard to the use of animals were approved by the Institutional Animal Care Committee of Nanjing First Hospital. All animal experiments were performed in accordance with the guidelines of decreasing the amount of suffering, pain, and discomfort of the experimental animals. Human clinical samples were obtained from patients who underwent total knee arthroplasties after informed consent and approval from the Ethics Committee of Nanjing First Hospital (Nanjing, China). All patients provided written informed consent prior to their inclusion within the study.
Consent for publication
All authors gave their consent for publication.
Availability of data and materials
The datasets used or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding
No
Authors' contributions
Jianchao Gui: Conceptualization, Methodology, Funding acquisition, Writing - Review & Editing
Zhaowei Yin: Validation, Investigation, Resources, Writing - Original Draft
Yan Feng and Chaoren Qin: Data Curation, Formal analysis
Shaowei Pan and Chen shi: Visualization, Formal analysis
Fu Guan and Jing Zhang: Resources, Investigation, Formal analysis, Visualization
Ziyi Yu: Conceptualization
Bin Liang: Supervision
All authors reviewed the manuscript.
Acknowledgements
Throughout the writing of this study, I have received a great deal of support and assistance.
I would first like to thank my supervisor, “Jianchao Gui”, whose expertise was invaluable in formulating the research questions and methodology. Your insightful feedback pushed me to sharpen my thinking and brought my work to a higher level.
I would particularly like to acknowledge my teammate members, “Yan Feng, Chaoren Qin Shaowei Pan, Chen Shi, Guanfu Wu, Jing Zhang, Ziyi Yu, Bin Liang”, for their wonderful collaboration and patient support and their valuable guidance throughout my studies. You provided me with the tools that I needed to choose the right direction and successfully complete my dissertation.
Finally, I could not have completed this study without the support of my wife, who devotes a lot of time to her family and allows me to rest my mind outside of my research.
- Wu Z, Yuan K, Zhang Q, Guo JJ, Yang H, Zhou F. Antioxidant PDA-PEG nanoparticles alleviate early osteoarthritis by inhibiting osteoclastogenesis and angiogenesis in subchondral bone. J Nanobiotechnol. BioMed Central; 2022;20:1–17.
- Xie W, Qi S, Dou L, Wang L, Wang X, Bi R, et al. Achyranthoside D attenuates chondrocyte loss and inflammation in osteoarthritis via targeted regulation of Wnt3a. Phytomedicine. 2023;111:154663.
- Arctiin-reinforced antioxidant microcarrier antagonizes osteoarthritis progression | Journal of Nanobiotechnology | Full Text [Internet]. [cited 2023 Feb 11]. Available from: https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-022-01505-7
- Yang H, Wang Z, Wang L, Li Y, Guo J, Yang X, et al. Scutellarin ameliorates osteoarthritis by protecting chondrocytes and subchondral bone microstructure by inactivating NF-κB/MAPK signal transduction. Biomedicine & Pharmacotherapy. 2022;155:113781.
- Auricular cartilage regeneration using different types of mesenchymal stem cells in rabbits | Biological Research | Full Text [Internet]. [cited 2023 Feb 11]. Available from: https://biolres.biomedcentral.com/articles/10.1186/s40659-022-00408-z
- Li S, Yang J, Sun J, Chen M. Adipose-Derived Mesenchymal Stem Cells Alleviate Hypertrophic Scar by Inhibiting Bioactivity and Inducing Apoptosis in Hypertrophic Scar Fibroblasts. Cells. 2022;11:4024.
- Bispo DSC, Jesus CSH, Romek K, Marques IMC, Oliveira MB, Mano JF, et al. An Intracellular Metabolic Signature as a Potential Donor-Independent Marker of the Osteogenic Differentiation of Adipose Tissue Mesenchymal Stem Cells. Cells. 2022;11:3745.
- Chen C-F, Hu C-C, Wu C-T, Wu H-TH, Chang C-S, Hung Y-P, et al. Treatment of knee osteoarthritis with intra-articular injection of allogeneic adipose-derived stem cells (ADSCs) ELIXCYTE®: a phase I/II, randomized, active-control, single-blind, multiple-center clinical trial. Stem Cell Res Ther. BioMed Central; 2021;12:1–12.
- Rochette L, Mazini L, Malka G, Zeller M, Cottin Y, Vergely C. The Crosstalk of Adipose-Derived Stem Cells (ADSC), Oxidative Stress, and Inflammation in Protective and Adaptive Responses. IJMS. 2020;21:9262.
- Billon N, Dani C. Developmental Origins of the Adipocyte Lineage: New Insights from Genetics and Genomics Studies. Stem Cell Rev and Rep. 2012;8:55–66.
- Lanthier N, Leclercq IA. Adipose tissues as endocrine target organs. Best Practice & Research Clinical Gastroenterology. 2014;28:545–58.
- Mochizuki T, Muneta T, Sakaguchi Y, Nimura A, Yokoyama A, Koga H, et al. Higher chondrogenic potential of fibrous synovium– and adipose synovium–derived cells compared with subcutaneous fat–derived cells: Distinguishing properties of mesenchymal stem cells in humans. Arthritis Rheum. 2006;54:843–53.
- Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obesity Reviews. 2010;11:11–8.
- Wickham MQ, Erickson GR, Gimble JM, Vail TP, Guilak F. Multipotent Stromal Cells Derived From the Infrapatellar Fat Pad of the Knee. Clinical Orthopaedics & Related Research. 2003;412:196–212.
- Lu V, Tennyson M, Zhang J, Khan W. Mesenchymal Stem Cell-Derived Extracellular Vesicles in Tendon and Ligament Repair—A Systematic Review of In Vivo Studies. Cells. 2021;10:2553.
- Sun H, Pratt RE, Hodgkinson CP, Dzau VJ. Sequential paracrine mechanisms are necessary for the therapeutic benefits of stem cell therapy. American Journal of Physiology-Cell Physiology. 2020;319:C1141–50.
- Ma J, Yong L, Lei P, Li H, Fang Y, Wang L, et al. Advances in microRNA from adipose-derived mesenchymal stem cell-derived exosome: focusing on wound healing. J Mater Chem B. 2022;10:9565–77.
- Wu J, Kuang L, Chen C, Yang J, Zeng W-N, Li T, et al. miR-100-5p-abundant exosomes derived from infrapatellar fat pad MSCs protect articular cartilage and ameliorate gait abnormalities via inhibition of mTOR in osteoarthritis. Biomaterials. 2019;206:87–100.
- Zhang Y, Wang X, Chen J, Qian D, Gao P, Qin T, et al. Exosomes derived from platelet-rich plasma administration in site mediate cartilage protection in subtalar osteoarthritis. J Nanobiotechnol. 2022;20:56.
- Liu Y, Zeng Y, Si H-B, Tang L, Xie H-Q, Shen B. Exosomes Derived From Human Urine–Derived Stem Cells Overexpressing miR-140-5p Alleviate Knee Osteoarthritis Through Downregulation of VEGFA in a Rat Model. Am J Sports Med. 2022;50:1088–105.
- Lei J, Jiang X, Li W, Ren J, Wang D, Ji Z, et al. Exosomes from antler stem cells alleviate mesenchymal stem cell senescence and osteoarthritis. Protein Cell. 2022;13:220–6.
- Li F, Xu Z, Xie Z, Sun X, Li C, Chen Y, et al. Adipose mesenchymal stem cells-derived exosomes alleviate osteoarthritis by transporting microRNA -376c-3p and targeting the WNT-beta-catenin signaling axis. Apoptosis [Internet]. 2022 [cited 2023 Feb 11]; Available from: https://link.springer.com/10.1007/s10495-022-01787-0
- Chen Y, Huang H, Zhong W, Li L, Lu Y, Si H. miR-140-5p protects cartilage progenitor/stem cells from fate changes in knee osteoarthritis. International Immunopharmacology. 2023;114:109576.
- Cai D, Zhang J, Yang J, Lv Q, Zhong C. Overexpression of FTO alleviates osteoarthritis by regulating the processing of miR-515-5p and the TLR4/MyD88/NF-κB axis. International Immunopharmacology. 2023;114:109524.
- Li Y, Xie W, Zheng Y, Li H, Wen Z, Wang C, et al. The miR-548d-5p/SP1 signaling axis regulates chondrocyte proliferation and inflammatory responses in osteoarthritis. International Immunopharmacology. 2022;110:109029.
- Garcia J, Wright K, Roberts S, Kuiper JH, Mangham C, Richardson J, et al. Characterisation of synovial fluid and infrapatellar fat pad derived mesenchymal stromal cells: The influence of tissue source and inflammatory stimulus. Sci Rep. 2016;6:24295.
- Klein-Wieringa IR, Kloppenburg M, Bastiaansen-Jenniskens YM, Yusuf E, Kwekkeboom JC, El-Bannoudi H, et al. The infrapatellar fat pad of patients with osteoarthritis has an inflammatory phenotype. Annals of the Rheumatic Diseases. 2011;70:851–7.
- Wei W, Rudjito R, Fahy N, Verhaar JAN, Clockaerts S, Bastiaansen-Jenniskens YM. THE INFRAPATELLAR FAT PAD FROM DISEASED JOINTS INHIBITS CHONDROGENESIS OF MESENCHYMAL STEM CELLS.
- Skubis-Sikora A, Sikora B, Witkowska A, Mazurek U, Gola J. Osteogenesis of adipose-derived stem cells from patients with glucose metabolism disorders. Mol Med. 2020;26:67.
- Liu S, Li R, Dou K, Li K, Zhou Q, Fu Q. Injectable thermo-sensitive hydrogel containing ADSC-derived exosomes for the treatment of cavernous nerve injury. Carbohydrate Polymers. 2023;300:120226.
- Hu N, Cai Z, Jiang X, Wang C, Tang T, Xu T, et al. Hypoxia-pretreated ADSC-derived exosome-embedded hydrogels promote angiogenesis and accelerate diabetic wound healing. Acta Biomaterialia. 2023;157:175–86.
- Zhao Y, Gong Y, Liu X, He J, Zheng B, Liu Y. The Experimental Study of Periodontal Ligament Stem Cells Derived Exosomes with Hydrogel Accelerating Bone Regeneration on Alveolar Bone Defect. Pharmaceutics. 2022;14:2189.
- Chen J, Wu J, Mu J, Li L, Hu J, Lin H, et al. An antioxidative sophora exosome-encapsulated hydrogel promotes spinal cord repair by regulating oxidative stress microenvironment. Nanomedicine: Nanotechnology, Biology and Medicine. 2023;47:102625.
- Sun Q, Yin W, Ru X, Liu C, Song B, Qian Z. Dual role of injectable curcumin-loaded microgels for efficient repair of osteoarthritic cartilage injury. Front Bioeng Biotechnol. 2022;10:994816.
- Xiong W, Lan Q, Liang X, Zhao J, Huang H, Zhan Y, et al. Cartilage-targeting poly(ethylene glycol) (PEG)-formononetin (FMN) nanodrug for the treatment of osteoarthritis. J Nanobiotechnol. 2021;19:197.
- Saornil JV, Sánchez Milá Z, Campón Chekroun AM, Baraja Vegas L, Vicente Mampel J, Frutos Llanes R, et al. Comparative Study of the Efficacy of Hyaluronic Acid, Dry Needling and Combined Treatment in Patellar Osteoarthritis—Single-Blind Randomized Clinical Trial. IJERPH. 2022;19:10912.
- Zhang J, Yong H, A S, Xu Q, Miao Y, Lyu J, et al. Structural Design of Robust and Biocompatible Photonic Hydrogels from an In Situ Cross-Linked Hyperbranched Polymer System. Chem Mater. 2018;30:6091–8.
- Yu H, Huang C, Kong X, Ma J, Ren P, Chen J, et al. Nanoarchitectonics of Cartilage-Targeting Hydrogel Microspheres with Reactive Oxygen Species Responsiveness for the Repair of Osteoarthritis. ACS Appl Mater Interfaces. 2022;14:40711–23.
- Xu H, Sun M, Wang C, Xia K, Xiao S, Wang Y, et al. Growth differentiation factor-5–gelatin methacryloyl injectable microspheres laden with adipose-derived stem cells for repair of disc degeneration. Biofabrication. 2020;13:015010.
- Zhao S, Wen H, Ou Y, Li M, Wang L, Zhou H, et al. A new design for living cell-based biosensors: Microgels with a selectively permeable shell that can harbor bacterial species. Sensors and Actuators B: Chemical. 2021;334:129648.
- Zhang J, Qin Y, Pambos OJ, Zhang J, Chen S, Yu Z, et al. Microdroplets confined assembly of opal composites in dynamic borate ester-based networks. Chemical Engineering Journal. 2021;426:127581.
- Hootman JM, Helmick CG, Barbour KE, Theis KA, Boring MA. Updated Projected Prevalence of Self-Reported Doctor-Diagnosed Arthritis and Arthritis-Attributable Activity Limitation Among US Adults, 2015-2040: PROJECTED PREVALENCE OF ARTHRITIS IN THE US, 2015-2040. Arthritis & Rheumatology. 2016;68:1582–7.
- Rannou F, Pelletier J-P, Martel-Pelletier J. Efficacy and safety of topical NSAIDs in the management of osteoarthritis: Evidence from real-life setting trials and surveys. Seminars in Arthritis and Rheumatism. 2016;45:S18–21.
- Tiku ML, Sabaawy HE. Cartilage regeneration for treatment of osteoarthritis: a paradigm for nonsurgical intervention. Therapeutic Advances in Musculoskeletal. 2015;7:76–87.
- Meng P, Zhang F, Zhang Y, Wei H, Tan S, Guo X, et al. ADAMTS4 and ADAMTS5 may be considered as new molecular therapeutic targets for cartilage damages with Kashin-Beck Disease. Medical Hypotheses. 2020;135:109440.
- Verma P, Dalal K. ADAMTS-4 and ADAMTS-5: Key enzymes in osteoarthritis. J Cell Biochem. 2011;112:3507–14.
- Tortorella MD, Malfait A-M, Deccico C, Arner E. The role of ADAM-TS4 (aggrecanase-1) and ADAM-TS5 (aggrecanase-2) in a model of cartilage degradation. Osteoarthritis and Cartilage. 2001;9:539–52.
- Karsenty G. An Aggrecanase and Osteoarthritis. N Engl J Med. 2005;353:522–3.
- Latourte A, Richette P. Inhibition of ADAMTS-5: the right target for osteoarthritis? Osteoarthritis and Cartilage. 2022;30:175–7.
- Han C, Zhou J, Liu B, Liang C, Pan X, Zhang Y, et al. Delivery of miR-675 by stem cell-derived exosomes encapsulated in silk fibroin hydrogel prevents aging-induced vascular dysfunction in mouse hindlimb. Materials Science and Engineering: C. 2019;99:322–32.
- Takahashi Y, Nishikawa M, Shinotsuka H, Matsui Y, Ohara S, Imai T, et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. Journal of Biotechnology. 2013;165:77–84.
- Imai T, Takahashi Y, Nishikawa M, Kato K, Morishita M, Yamashita T, et al. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. Journal of Extracellular Vesicles. 2015;4:26238.
- Zhang S, Chu WC, Lai RC, Lim SK, Hui JHP, Toh WS. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthritis and Cartilage. 2016;24:2135–40.
- Brennan MÁ, Layrolle P, Mooney DJ. Biomaterials Functionalized with MSC Secreted Extracellular Vesicles and Soluble Factors for Tissue Regeneration. Adv Funct Mater. 2020;30:1909125.
- Nikravesh N, Davies OG, Azoidis I, Moakes RJA, Marani L, Turner M, et al. Physical Structuring of Injectable Polymeric Systems to Controllably Deliver Nanosized Extracellular Vesicles. Adv Healthcare Mater. 2019;8:1801604.
- Mol EA, Lei Z, Roefs MT, Bakker MH, Goumans M, Doevendans PA, et al. Injectable Supramolecular Ureidopyrimidinone Hydrogels Provide Sustained Release of Extracellular Vesicle Therapeutics. Adv Healthcare Mater. 2019;8:1900847.
- Han Y, Yang J, Zhao W, Wang H, Sun Y, Chen Y, et al. Biomimetic injectable hydrogel microspheres with enhanced lubrication and controllable drug release for the treatment of osteoarthritis. Bioactive Materials. 2021;6:3596–607.
- Lin J, Chen L, Yang J, Li X, Wang J, Zhu Y, et al. Injectable Double Positively Charged Hydrogel Microspheres for Targeting‐Penetration‐Phagocytosis. Small. 2022;18:2202156.
- Seong Y-J, Lin G, Kim BJ, Kim H-E, Kim S, Jeong S-H. Hyaluronic Acid-Based Hybrid Hydrogel Microspheres with Enhanced Structural Stability and High Injectability. ACS Omega. 2019;4:13834–44.
- Gerwin N, Hops C, Lucke A. Intraarticular drug delivery in osteoarthritis☆. Advanced Drug Delivery Reviews. 2006;58:226–42.
- Yang J, Zhu Y, Wang F, Deng L, Xu X, Cui W. Microfluidic liposomes-anchored microgels as extended delivery platform for treatment of osteoarthritis. Chemical Engineering Journal. 2020;400:126004.
- Lin J, Xue J, Xu Q, Liu Z, Zhao C, Tang J, et al. In situ-crosslinked hydrogel-induced experimental glaucoma model with persistent ocular hypertension and neurodegeneration. Biomater Sci. 2022;10:5006–17.
No competing interests reported.
- S1.jpg
Fig S1. The IVIS images of mice after injection of the ExosScAT, ExosScAT-99b-3p, HMPs@Exos, HMPs@ExosScAT-99b-3p for specific time points.
- S2.jpg
Fig S2. Histological evaluations of mice organs (Lung, Liver, Kidney, Heart, Spleen) on day 7 after treatment with Saline, ExosScAT, ExosScAT-99b-3p, HMPs@Exos, HMPs@ExosScAT-99b-3p. Scale bar=50 μm.
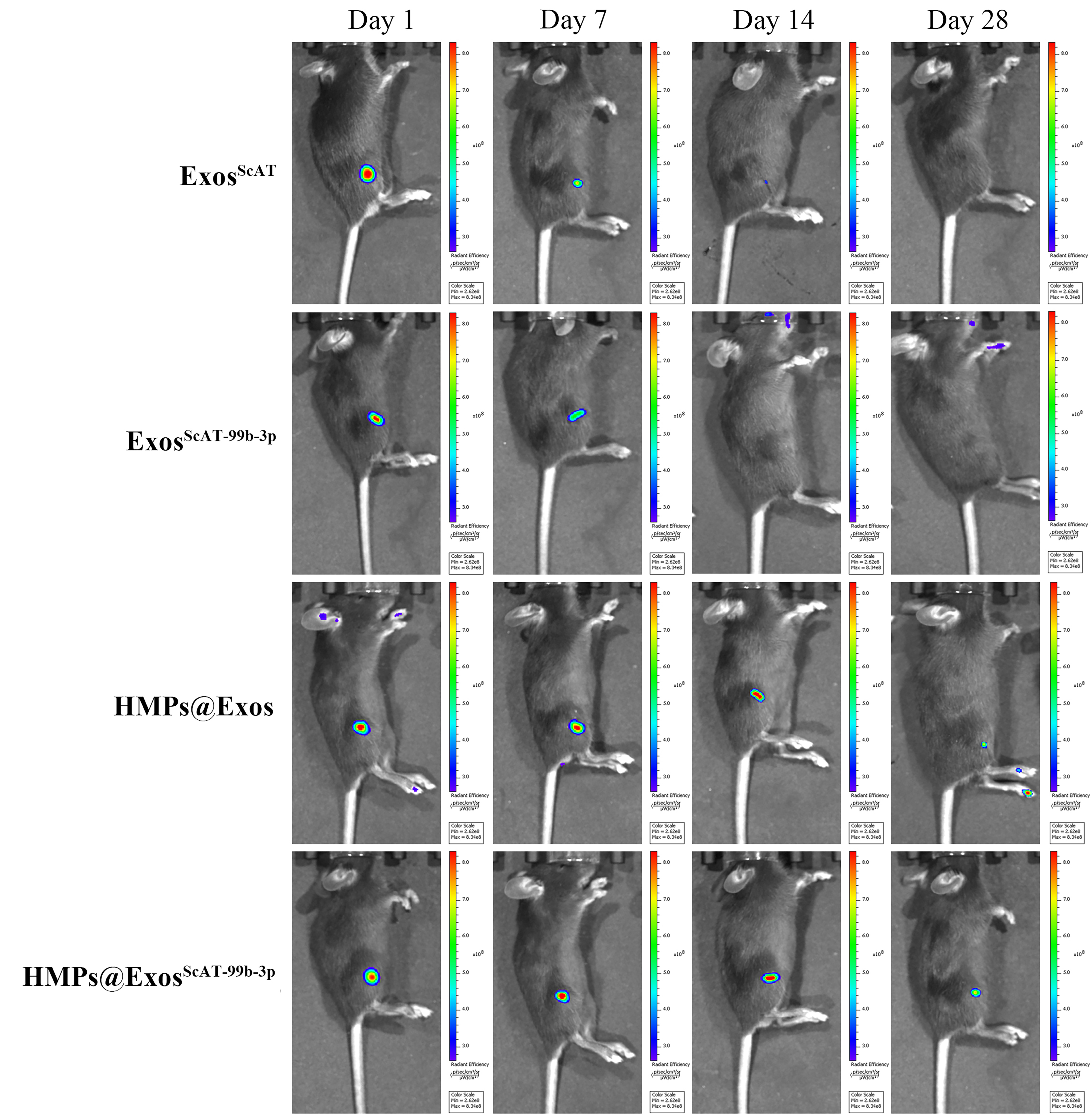{kind=link}
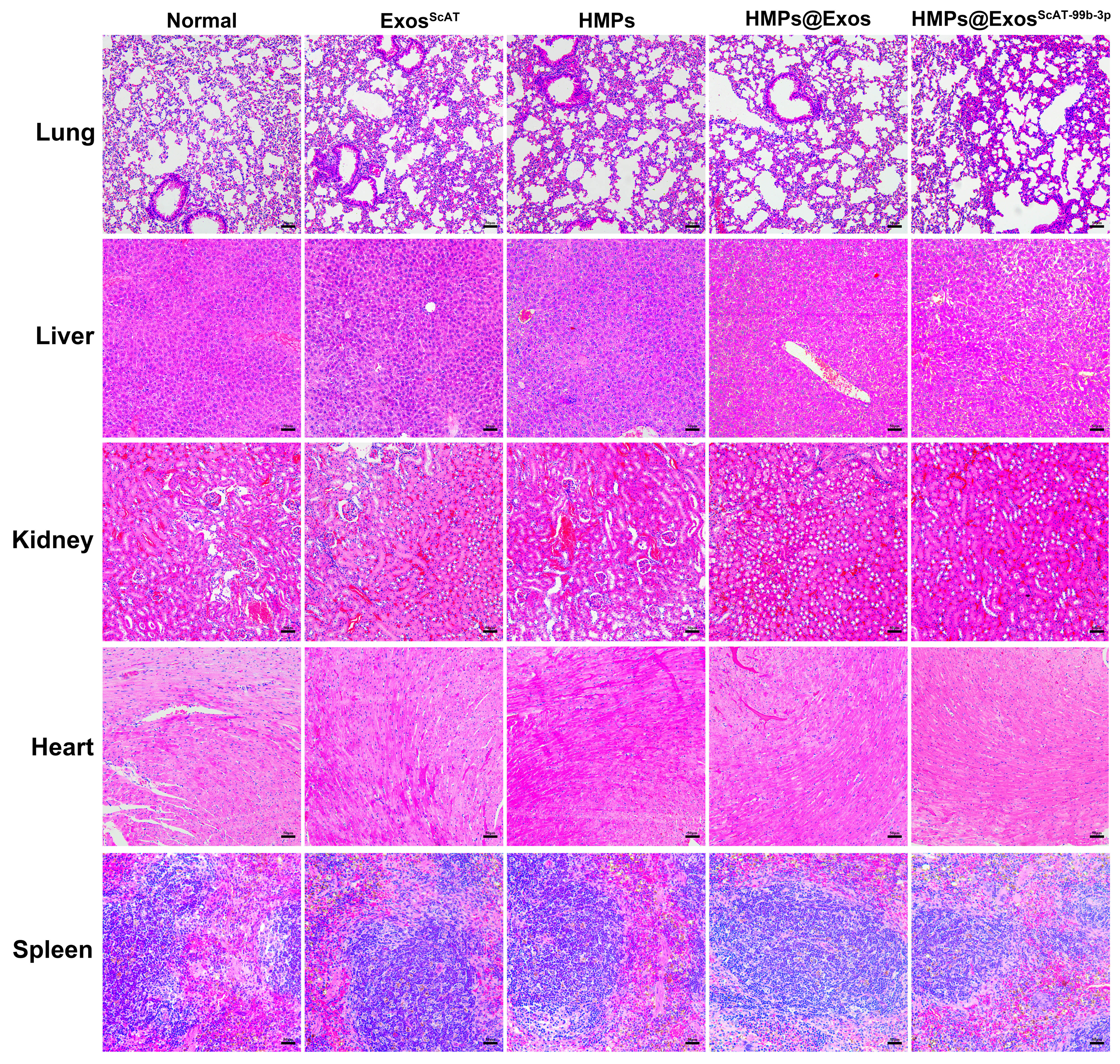{kind=link}