Design
ProCCard is a multicenter, prospective, randomized, controlled, single-blinded, two-arm study comparing a bundle of care combining five techniques of cardioprotection to a conventional approach in cardiac surgery with CPB. The trial is performed in accordance with the declaration of Helsinki (revised version, 2013), the European Guidelines for Good Clinical Practice (revised version, 2016), and the French laws.
Partners
Patients are recruited in six French institutions (details in Table 1). The study sponsor is the Direction of Clinical Research of the Hospices Civils de Lyon, a public academic institution in France. The Clinical Investigation Center of Lyon, an academic research organization within the Hospices Civils de Lyon (Inserm 1407), provides the methodological support, coordinates the trial and also collects the trial data. All the analyses will be performed by the Biostatistics Department at the Hospices Civils de Lyon which also provides methodological support to the study. The study is supported by the French Clinical Research Program 2016 (Programme Hospitalier de Recherche Clinique).
Study population
Screening and inclusion
Patients are screened during the preoperative visit by the anesthesiologist, informed, and included after providing written consent.
Inclusion criteria
Patients scheduled for aortic valve surgery, with or without coronary artery bypass, older than eighteen years, are eligible for enrollment.
Exclusion criteria
- Emergency surgery,
- Redo surgery,
- Preoperative treatment with nicorandil (an adenosine triphosphate-sensitive potassium channel opener), sulfonylurea or repaglinide (two adenosine triphosphate-sensitive potassium channel blockers),
- Preoperative acute circulatory insufficiency justifying catecholamine support or mechanical circulatory assistance,
- Severe chronic renal insufficiency (glomerular filtration rate < 30 ml/min),
- Severe chronic liver disease (spontaneous international normalized ratio > 2),
- Severe respiratory insufficiency (forced expiratory volume in one second < 40% theoretical value),
- Acute coronary syndrome less than seven days old,
- Current infections,
- Peripheral arterial disease at upper limbs,
- Any other surgical procedure associated to aortic valve surgery (combined valve surgery, Morrow’s myotomy…).
Perioperative procedure
Standard intraoperative monitoring consists of a 5-lead electrocardiogram, frontal electroencephalography (BIS-monitor A2000Ò; Aspect Medical Systems, Norwood, MA), pulse oximetry, peripheral arterial catheter, central pressure monitoring, capnography and vesical temperature probe.
Intravenous anesthesia is induced with either etomidate or propofol, sufentanil or remifentanil (to the discretion of the investigator), and cisatracurium. The maintenance of anesthesia depends on the experimental group assignation. After systemic heparinisation (300 IU/kg, activated clotting time > 400 s), the ascending aorta and right atrium are cannulated. A standard CPB with a disposable hollow-fiber membrane oxygenator is started with a target output of 2.4 l/min/m² of body surface area. Surgery is performed under mild hypothermia (> 35°C). After aortic cross-clamping, cardioplegia is achieved with crystalloid or blood solution, according to local protocols. After aortic unclamping, the heart is defibrillated if sinus rhythm does not resume spontaneously. Patients are transferred to the intensive care unit (ICU) and extubated when pressure support ventilation is tolerated.
Experimental protocol
Randomization
Written informed consent is obtained for all patients before inclusion. Patients are randomly assigned to either the intervention or control group. Computer-generated randomized lists are drawn up by the Biostatistics Department of the Hospices Civils de Lyon before the start of the study. The randomization is stratified by i) centers and ii) individual need to perform coronary bypass during aortic valve surgery. The allocation is implemented in the electronic case report form.
Treated group
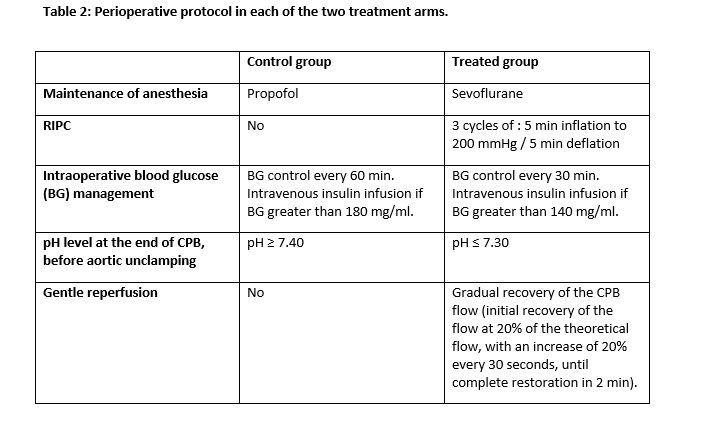
In the treated group, patients receive a bundle of care combining five techniques of cardioprotection (see Table 2):
(1) After intravenous induction, anesthesia is maintained during surgery with sevoflurane to induce both preconditioning and postconditioning. During CPB, sevoflurane is administered through the heart-lung machine only in two institutions (Lyon University Hospital and Arnault Tzanck Institute). In the four remaining centers, because the device for administering sevoflurane is not available on heart-lung machine, propofol is administered during CPB.
(2) RIPC is applied using a RiesterÒ pneumatic tourniquet placed on the upper arm (Riester Company, 72417 Jungingen, Germany). Three cycles of 5 minutes inflation to 200 mmHg / 5 minutes deflation are performed after anesthesia induction and before skin incision.
(3) Intraoperative blood glucose management involves the initiation of insulin infusion when blood glucose, measured every 30 minutes, is greater than 140 mg/dl.
(4) During CPB, 5 minutes before aortic unclamping, blood gases are adjusted, by limiting the flow of air/oxygen mixture on the heart-lung machine, in order to obtain an arterial blood pH lower than or equal to 7.30, and consequently to limit the “pH paradox” phenomenon occurring at the time of reperfusion [11-12].
(5) Just after aortic unclamping, a “gentle reperfusion” protocol is applied, consisting of a gradual recovery of the CPB flow (initial recovery of the flow at 20% of the theoretical flow, with an increase of 20% every 30 seconds, until a complete restoration of the CPB flow in 2 minutes) to limit reperfusion injuries.
Control group
In the control group, patients are managed according to a standard of care (see Table 2):
(1) Anesthesia is maintained with continuous infusion of propofol during surgery (halogenated volatile anesthetics are not allowed in this group).
(2) Intraoperative blood glucose management involves the initiation of insulin infusion when blood glucose, measured every 60 minutes, is greater than 180 mg/dl.
(3) During CPB, 5 minutes before aortic unclamping, blood gases are adjusted by modifying the flow of air/oxygen mixture on the heart-lung machine, to obtain an arterial blood pH higher than or equal to 7.40.
(4) Just after aortic unclamping, the theoretical CPB flow is restored at the earliest, according to the clinical tolerance of the patient (no gentle reperfusion).
Outcomes
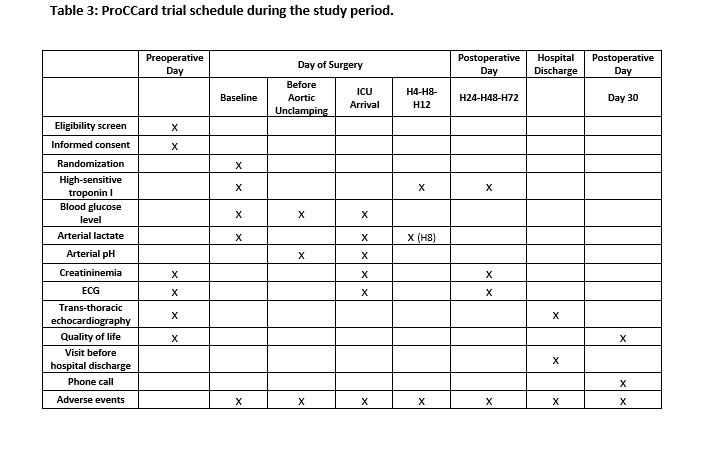
The primary outcome is the postoperative 72-h area under the curve (AUC) for high-sensitive cardiac troponin I (hsTnI) release. Blood samples are collected after induction of anesthesia (Baseline) and 4, 8, 12, 24, 48, and 72 h after aortic unclamping (see Table 3). All samples are frozen carefully and sent to a centralized laboratory for measurement (see below).
The secondary outcomes are: hsTnI level (peak and 24 h after aortic unclamping), extubation time, length of stay in ICU and hospital, Index Gravity Score (IGS II, the scoring system measuring the severity of disease for patients admitted to ICU), the 3-level version of EQ-5D (EQ-5D-3L) health status score (comparison between preoperative and 30 day postoperative), and the need for catecholaminergic support [13]. Major adverse events occurring during 30 days postoperative are collected and reported as follows: all-cause of death, serious infection requiring antibiotic therapy, major bleeding requiring transfusion of ≥ 5 U packed red blood cells or surgical intervention, respiratory insufficiency, stroke, arrhythmia requiring medical therapy.
Data collection
Baseline data
The following data are collected in the preoperative period: initial valvular aortic pathology requiring surgical correction, concomitant pathology (hypertension, dyslipidemia, diabetes, stroke, atrial fibrillation, chronic obstructive pulmonary disease, smoking) and medications (β-blockers, nitrates, statins, Ca²+ channel blockers, angiotensin-converting enzyme inhibitor, angiotensin II receptor blocker, diuretics, platelet inhibitors, insulin, oral antidiabetic drugs, amiodarone, heparin, oral anticoagulant therapy), age, sex, body mass index, NYHA class, ASA class, Logistic EuroSCORE I, blood creatinine level, blood hemoglobin level, electrocardiogram, EQ-5D-3L health status score, echocardiography (left ventricular (LV) ejection fraction, LV mass index calculated using: LV end-diastolic diameter, LV end-diastolic posterior wall thickness, LV end-diastolic interventricular wall thickness).
Intraoperative data
During surgery, the following data are collected: CPB duration (min), aortic cross-clamping duration (min), type and volume of cardioplegia (ml), surgical procedure performed, baseline hsTnI level, baseline lactate level, total iv sufentanil / remifentanil (µg), total iv propofol (mg), total iv insulin (IU), catecholaminergic treatment, blood glucose concentrations during the operative period, arterial blood gas harvested 5 min before aortic unclamping.
Postoperative data
The following data are also collected (see Table 3): blood lactate (ICU arrival, and 8 h after aortic unclamping) and creatinine (ICU arrival, 24, 48, and 72 h after aortic unclamping) levels, electrocardiogram (ICU arrival, 24, 48, and 72 h after aortic unclamping), extubation time, SAPS II, length of stay in ICU and hospital, echocardiography at 5 day postoperative (LV ejection fraction, LV mass index), EQ-5D-3L health status score at 30 day postoperative (phone call), any adverse event occurring during this period.
Centralized analysis of hsTnI
Blood samples for the analysis of hsTnI are collected in each institution, centrifuged at 2000 g for 10 min. The supernatant is removed and stored in a freezer (-80°C) before transfer for centralized analysis to the Biochemical Laboratory at the Lyon University Hospital (ARCHITECT STAT Troponin I; Abbott, North Chicago, IL).
Sample size
The sample size calculation was performed according to hypotheses stemming from a previous study carried out in the department of cardiac anesthesia of the same facility [6]. One hundred patients per arm would ensure an 80% power of detecting a decrease in the AUC of hsTnI release within the three first postoperative days (AUC of 242 in the control arm and 169 in the treatment arm) considering a common standard deviation of 183 and a two-sided Student test. Assuming a 5% rate of missing data, there should be 105 patients in each arm; thus, 210 patients to be included in the study.
Statistical analyses
The main analysis will be carried out within the intention-to-treat (ITT) population. The ITT population is defined as all patients included in the study whatever the selection criteria, the treatment strategy, or the adequacy for evaluation. All missing data necessary for the calculation of the outcome criterion (AUC of serum level of hsTnI within three days after surgery) will be imputed. The causes and contexts of missingness will orient the choice of the imputation method. The main analysis will compare the AUC of serum level of hsTnI within three days after surgery between the treatment and the control arm using a mixed-effect generalized linear model. According to the recommendations of the international conference on harmonization of technical requirements for the registration of pharmaceuticals for human use (ICH), each variable used for the stratification of the randomization will be introduced in the model as a random effect or fixed effect. Variable “arm” will be introduced as a fixed effect. Statistical significance on two-sided Wald test will be set at 0.05.
The same analysis will be carried out on the per-protocol (PP) population. The PP population is defined as all patients of the ITT population with no major deviation from the study protocol. This analysis will consider the actually applied strategy. A sensitivity analysis will be carried out after excluding all patients who experienced, within three days after surgery, any event able to bias the analysis of the main outcome. The main outcome will be also analyzed using a mixed-effect generalized linear model to investigate the effect of patient management through considering various practices likely to influence hsTnl kinetics; especially, glycemic and lactic acidosis monitoring.
All secondary outcomes will be summarized, per arm, according to their nature (qualitative or quantitative). The peak of serum level of hsTnI and the level of hsTnI at 24 hours after aortic unclamping will be analyzed in the same way as the main outcome. The duration of mechanical ventilation and the duration of stay in the ICU will be similarly analyzed either after log-transformation of the values or using a survival analysis technique (time-to-event analysis of the time to mechanical ventilation discontinuation or the time to discharge from hospital, respectively). A frailty Cox model may be used to allow for the variables used for stratification. The length of hospital stay will be analyzed with a Cox frailty model adjusted for the variables used for stratification. The IGS II will be analyzed in the same way as the main outcome after checking the score distribution. If necessary, an adequate score transformation may be applied.
The need for catecholaminergic support will be analyzed with a logistic model adjusted for the variables used for stratification.
The occurrence of at least one postoperative complication will be analyzed with a logistic model adjusted for the variables used for stratification. The complications considered are: post-operative hemorrhage, need for postoperative blood transfusion, the need for revision surgery for bleeding, the occurrence of respiratory insufficiency requiring intubation or not, postoperative infection, neurological disorder, and complete arrhythmia by atrial fibrillation. The postoperative adverse events will be analyzed in the safety population. The safety population is defined as all patients of the ITT population who received any treatment of the actually applied strategy. The proportions of patients concerned by the secondary outcomes will be calculated by arm and type of complication.
The score of EQ-5D-3L will be analyzed using a mixed linear regression model that will include a variable for “time” (at 30 days after surgery vs. before surgery), a variable for “group”, and an interaction between these two variables. This model will be also adjusted for the variables used for stratification. The coefficient of interaction will be tested to compare the arms. The score distribution will be examined for floor and/or ceiling effects. If necessary a Tobit model will be used. Survival will be analyzed with a Cox model adjusted for the variables used for stratification.
Regulatory issues
The study has been approved by the French National Agency for Medicines and Health Products Safety (Agence Nationale de Sécurité du Médicament et des Produits de Santé, 93285 Saint-Denis, France) on August 16, 2017 (ID-RCB: 2017-A00694-49) and by an independent ethics committee (Comité de Protection des Personnes, Ile-de-France VII, 94270 Le Kremlin Bicêtre, France) on September 13, 2017. The study was registered on ClinicalTrials.gov on July 26, 2017 (NCT03230136). Central ethical approval has been confirmed from Comité de Protection des Personnes (ref approval N° 2017-A00694-49) and we will not begin recruiting at other centers in the trial until local ethical approval has been obtained. Patient data are collected anonymously on the electronic platform. All severe adverse events are documented in the electronic case report form and declared to the Pharmacovigilance Risk Assessment Committee (Hospices Civils de Lyon). An independent Data Safety Monitoring Board including three external experts (Pr Claude Girard, anesthesiologist, Dijon, France, Pr Julien Amour, anesthesiologist, Paris, France and Dr. Remy Morello, biostatistician, Caen, France) conducts a safety monitoring.
{kind=link}
{kind=link}
{kind=link}