Catalysts Synthesis and Characterization
The synthesis of iron catalysts using two MOF precursors (ZIF-8 and MOF-5) is illustrated in Fig. 1a. Fe(NO3)3, a smaller Fe salt, is used as the Fe source, compared to literature’s Fe(acac)3 [27]. The catalyst prepared using the ZIF-8 precursor after pyrolysis at 800 oC contains ultra-low loading of Fe (< 0.1%) vs. 2.16% in Li’s paper [27] (Table S1). As Fe coordinates with H2BDC but not 2-MI, saturated adsorption of Fe3+ onto ZIF-8 using 2-MI is recommended to achieve low Fe loading even upon adding excessive Fe precursor. To tune the Fe loading in Fe-ZIF catalysts, a two-step route is employed, while similar performance is achieved as the one-step route at the same Fe loading (see details in methods section).
TEM images of the ZIF-8 precursor (Figure S1) and Fe-ZIF-8-800 catalyst (Fig. 1c) reveal a uniform lamellar structure of ca. 160 nm ZIF-8 matrix that is well preserved during pyrolysis. Elemental mapping (Fig. 1f) suggests even dispersion of iron in Fe-ZIF-8-800 catalyst, and ICP-AES analysis shows a loading as low as < 0.1 wt%. In contrast, pyrolysis of MOF-5 leads to irregular particles (Fig. 1d), indicating the collapse of initial MOF-5 structure during pyrolysis. Characteristic peaks of ZnO (JCPDS 80–0075) and an extremely weak peak of the (110) facet of metallic iron (JCPDS 87–0721) are observed in the XRD pattern of Fe-MOF-5-800 catalyst. The MOF-5 affords a Fe loading of 5.73% (Table S1). The amorphous carbon peaks in Fe-ZIF-8-800 and ZIF-8-800 and the absence of ZnO and metallic Fe peaks in the XRD patterns of the ZIF-based catalyst (Fig. 1g) suggest that Zn and Fe are in a different state in MOF-5 and ZIF-8 catalysts.
The states of Zn and N were investigated via XPS. The binding energy of Zn in Fe-ZIF-8-800 is slightly higher than in ZIF-8-800, suggesting electron withdrawal from Zn to Fe (Fig. 1i). Zn’s binding energy in MOF-5 shifts to higher values, correlating to ZnO observed in the XRD pattern (Fig. 1g). Thus, Zn in ZIF-8 may be in ZnN4, as further confirmed by EXAFS (Fig. 2f). The fraction of pyrrolic N (the most possible N state in Fe-N3 moiety in Fig. 2g [28–30]) in Fe-ZIF-8-800 is 13.1 at%, higher than Fe-ZIF catalysts pyrolyzed at lower temperatures, and slightly lower than ZIF-8-800 (Fig. 1j and Table S5). Complete loss of Zn and N occurs upon pyrolysis at 1000 oC.
X-ray absorption near-edge structure (XANES) and Fourier transforms (FTs) of EXAFS spectra provide insights into the state of Fe even at low fractions as aberration-corrected transmission electron microscopy (Figure S8) cannot distinguish Fe and Zn atoms due to approximate atomic number [31]. Fe atoms in the Fe-ZIF-8-800 catalyst are oxidized (Fig. 2a), and the Fe-N length is ca. 1.4 Å (Fig. 2c). Trace Fe has limited influence on the oxidation state of Zn, leading to a nearly indistinguishable change in the XANES spectra of Zn K-edge (black and red lines in Fig. 2b). Only one main peak of Zn-N coordination is observed in the ZIF-8 catalysts (Fig. 2d). Wavelet transform spectra further confirm that Fe and Zn atoms are well dispersed without aggregation (Fig. 2i), whereas Fe-Fe and Zn-Zn scatterings are observed at about 6 Å in the Fe-MOF-5-800 catalyst (Figure S9g). The Fe-N coordination number in Fe-ZIF-8-800 catalyst is 3.7 (Table S9), suggesting a mixture of FeN3 and FeN4 structures (Figs. 2e and S10). The coordination number of Zn-N in Fe-ZIF-8-800 is similar to ZIF-8-800 (3.89 vs. 4.1) (Table S9), further confirming the limited influence of trace Fe on the structure of Zn (Fig. 2f).
Computed XANES spectra of metal-NxCy moieties [28–30] (metal = Fe or Zn) assist identifying the number and type of N ligands coordinated with the transition metal at the Fe-Nx and Zn-Nx coordination sites in Fe-ZIF-8-800 and ZIF-8-800. The porphyrin-based metal-NxCy moieties, in which the metal is coordinated to pyrrolic N atoms (plN), e.g., Fe(I)-N3C10, Zn(II)-N4C12, and Fe(II)-N4C12 (Figs. 2g, h, and S11a), reproduce the experimental spectra the best. When pyridinic N is involved in Fe-N coordination (Figure S12), the edge and post-edge experimental features were not reproduced. Taken together, the EXAFS and XANES analyses suggest that the local structures of the Fe-Nx sites in Fe-ZIF-8-800 are described well by porphyrin-based moieties Fe(II)-N4C12 and Fe(I)-N3C10; and the Zn-Nx site of ZIF-8-800 by the Zn(II)-N4C12 moiety.
Catalytic Performance
Figure 3a shows the catalytic performance and TOF of various catalysts, illustrating the importance of MOF precursors and Fe on reactivity. Some other effects like pyrolysis temperature or H donor were shown in Tables S10 and S11. Strikingly, Fe-ZIF-8-800 catalyst affords 99.6% FF conversion and 96.9% FA selectivity at 120°C in 6 h. This performance is comparable to state-of-the-art catalysts (Table S12) [25, 32–42], significantly better than our previous Fe catalysts, named Fe-phen/C-800 [25], and much better than other Fe catalysts under mild conditions (Table S13) [25, 43–49]. Fe-MOF-5-800 and ZIF-8-800 catalysts give lower FF conversion and FA selectivity and TOF of ca. 0.054 h–1, indicating that the MOF-5 precursor and absence of Fe lead to inferior catalytic activity. Catalyst precursors without pyrolysis, with or without Fe, give poor FA yields. In an 1 h, only a slight drop in FF conversion and FA yield are achieved.
The TOF of Fe catalysts is benchmarked in Fig. 3a. Clearly, the TOF of Fe-ZIF-8-800 catalyst (2435 h–1, calculated at 120 oC and 2 min with a conversion of 10.7%) is two to four orders of magnitude higher than other Fe-based catalysts. The excellent activity of the SA Fe underscores that ZIF catalysts have a unique active site.
The effect of the reaction temperature in the range of 80 to 160 oC is shown in Figure S13. Notably, the Fe-ZIF-8-800 catalyst affords a TOF of 2435 h–1 at 120 oC, 267 times higher than that of Fe-phen/C-800 catalyst [25] due to its highly active sites. At 160 oC, a 96.6% yield of FA is obtained in 0.5 h with complete conversion of FF over Fe-ZIF-8-800 catalyst vs. 36.5% yield with 42.9% conversion over Fe-phen/C-800 The major by-products are aldol condensation products of FF and acetone on Fe-ZIF-8-800, also observed over the Fe-phen/C-800 catalyst [25]. At 80 oC in 3 h, the catalytic performance of Fe-ZIF-8-800 at a higher metal loading of 0.54 mol% is comparable to state-of-the-art catalysts (Table S12).
Reaction Mechanism
The linear correlation between Fe loading and specific reaction rate clearly indicates that SA Fe species are the active sites (Fig. 3b). Isotopic labeling experiments confirmed that CTH reaction proceeds via intermolecular hydride transfer, rather than metal-mediated hydrogenation, following the Meerwin-Ponndorf-Verley (MPV) mechanism over Fe-ZIF-8-800 and ZIF-8-800 because of a 1 amu mass spectrometry (MS) shift of the FA formed using 10% 2-propanol-d8 in t-butanol solution (Fig. 3c) [50].
To better understand the differences in catalytic performance of Fe-ZIF-8-800 and ZIF-8-800, we performed DFT calculations for the MPV mechanism on periodic models of Fe(I)-plN3, Fe(II)-plN4, and Zn(II)-plN4 (Figure S14), consisting of Fe(I)-N3C10, Fe(II)-N4C12, and Zn(II)-N4C12 moieties confirmed from the XANES simulations (Figs. 2g and h).
In a typical MPV reduction of aldehydes over acid-base site pairs of Lewis acid metal-substituted zeolites (e.g., Sn-beta) [51, 52], the proton of the alcohol is abstracted by the basic site while the Lewis acid center coordinates the conjugate base of the alcohol (alkoxide ion) and the aldehyde in an octahedral geometry. The ensuing direct hydride transfer from the alkoxide ion to the aldehyde proceeds via a six-member-ring transition state. On Fe(II)-plN4 and Zn(II)-plN4, however, the Fe and Zn atoms cannot coordinate both reactants at the same time due to steric hindrance from the rigid geometry of the metal-N4 site. Nevertheless, the reduction of FF can proceed on both Fe(II)-plN4 and Zn(II)-plN4 by a non-typical MPV mechanism. We have identified two such pathways, one with FF and another with isopropanol (IPA) coordinated to the metal center, respectively referred to as pathway 1 (P1) and pathway 2 (P2) in Fig. 3d. In both cases, the reduction proceeds through a single transition state in which the proton and the α-hydride transfer from IPA to FF in a coordinated manner, leading to FA and acetone (ACE). The optimized geometries of the intermediates and transition states are shown in Figures S15 and S16. The corresponding Gibbs free energy profiles are shown in Figs. 3f and g. The energy span of P2 (ca. 1.4 eV for both Fe(II) and Zn(II)) is somewhat greater than that of P1 (ca. 1.2 eV for both Fe(II) and Zn(II)), mainly because when IPA coordinates to the metal, FF physisorbs on the carbon support more strongly than IPA does when the situation is reversed (viz. FF coordinated to the metal) but without a commensurate stabilization of the corresponding transition state. Irrespective of the non-typical MPV pathway, however, the similar Fe(II)-plN4 and Zn(II)-plN4 activities are in stark contrast to our experimental data, which show a very low activity of Zn, and suggest that Fe(II)-plN4 might not be the active site responsible for the higher activity of Fe-ZIF-8-800, as shown in Fig. 3a and Table S10.
In contrast, the Fe(I)-plN3 active site model can support a typical MPV mechanism, whereby IPA binding to the Fe(I) atom and deprotonation by a vicinal N-atom is followed by FF coordination to the Fe(I) atom. The FF binding is enabled by the protonation of the N-ligand which weakens the respective Fe-N bond and lets the Fe atom relieve the strain associated with the FF binding by being pulled out of the plane of the support (see optimized geometries in Figure S17). This pathway (P3) is illustrated in detail in Fig. 3e and the corresponding free energy profile in Fig. 3h. The deprotonation of IPA is facile, requiring 0.28 eV (TS1 in Fig. 3h). The rate-limiting step is the α-hydride transfer from the alkoxide ion to the carbonyl C atom of FF via a six-member-ring transition state (TS2). This step requires 0.87 eV of activation energy (intrinsic) and also determines the overall energy span on this pathway, which is significantly lower than those calculated for the Fe(II)-plN4 and Zn(II)-plN4 models. The reaction is completed by a proton backdonation from the pyrrolic N atoms to the surface furoxy species, FF*. Microkinetic simulation of the reactions on Fe(I)-plN3, Fe(II)-plN4, and Zn(II)-plN4 showed that the TOF of Fe(I)-plN3 is higher than those of Zn(II)-plN4 and Fe(II)-plN4 by six orders of magnitude (Table S14), suggesting that Fe(I)-plN3 is mostly responsible for the high catalytic activity of Fe-ZIF-8-800 compared to ZIF-8-800.
We note, in passing, that for completeness we also investigated the non-typical MPV pathways P1 and P2 on Fe(I)-plN3. However, the energy spans were 1.19 eV and 1.25 eV for P1 and P2, respectively (Figure S18), namely, only slightly lower than those on Zn(II)-plN4, which, once again, does not explain the higher activity of Fe-ZIF-8-800 relative to ZIF-8-800.
The higher activity of Fe(I)-plN3 relative to the other Fe-sites considered here should be attributed to the anchoring of the Fe atom to a defect site. The resulting three-coordinate metal site in the defective structure promotes the P3 pathway by enabling the FF binding on the Fe(I) center after the deprotonation of IPA. In contrast, Fe(II)-plN4 only supports the relatively unfavorable P1 and P2 pathways because it cannot coordinate both reactants simultaneously. The reasons can be qualitatively understood by considering the respective crystal field splitting diagrams [53, 54]. As shown in Figure S19a, coordination of FF to the Fe(I) center of the trigonal complex depIPA_H* leads to the tetrahedral complex depIPA_H_FF* which is no less stable than depIPA_H* on account of the partially occupied \({d}_{xy}\), \({d}_{xz}\) and \({d}_{yz}\) orbitals lying lower in energy than the \({d}_{{x}^{2}-{y}^{2}}\) and \({d}_{xy}\) orbitals of the trigonal complex. On the other hand, if the square pyramidal Fe(II) center of Fe(II)-plN4 were allowed to bind a second reactant, the uncommon “ridge-tent” looking complex would form, with the two reactants at the ridge over the square planar Fe-N4 complex (Figure S19b). This would be an unstable complex because the crystal field of this “tent” configuration would be destabilized on account of the four electrons in three orbitals (\({d}_{{z}^{2}}, {d}_{xz}\) and \({d}_{yz}\)) above the barycenter of the field, compared to only one unpaired electron in an orbital (\({d}_{{z}^{2}}\)) above the barycenter of the square pyramidal complex with only one of the two reactants as a ligand. This unfavorable electronic configuration precludes coordination of both IPA and FF to Fe(II), inhibiting the P3 pathway.
Substrate Effect
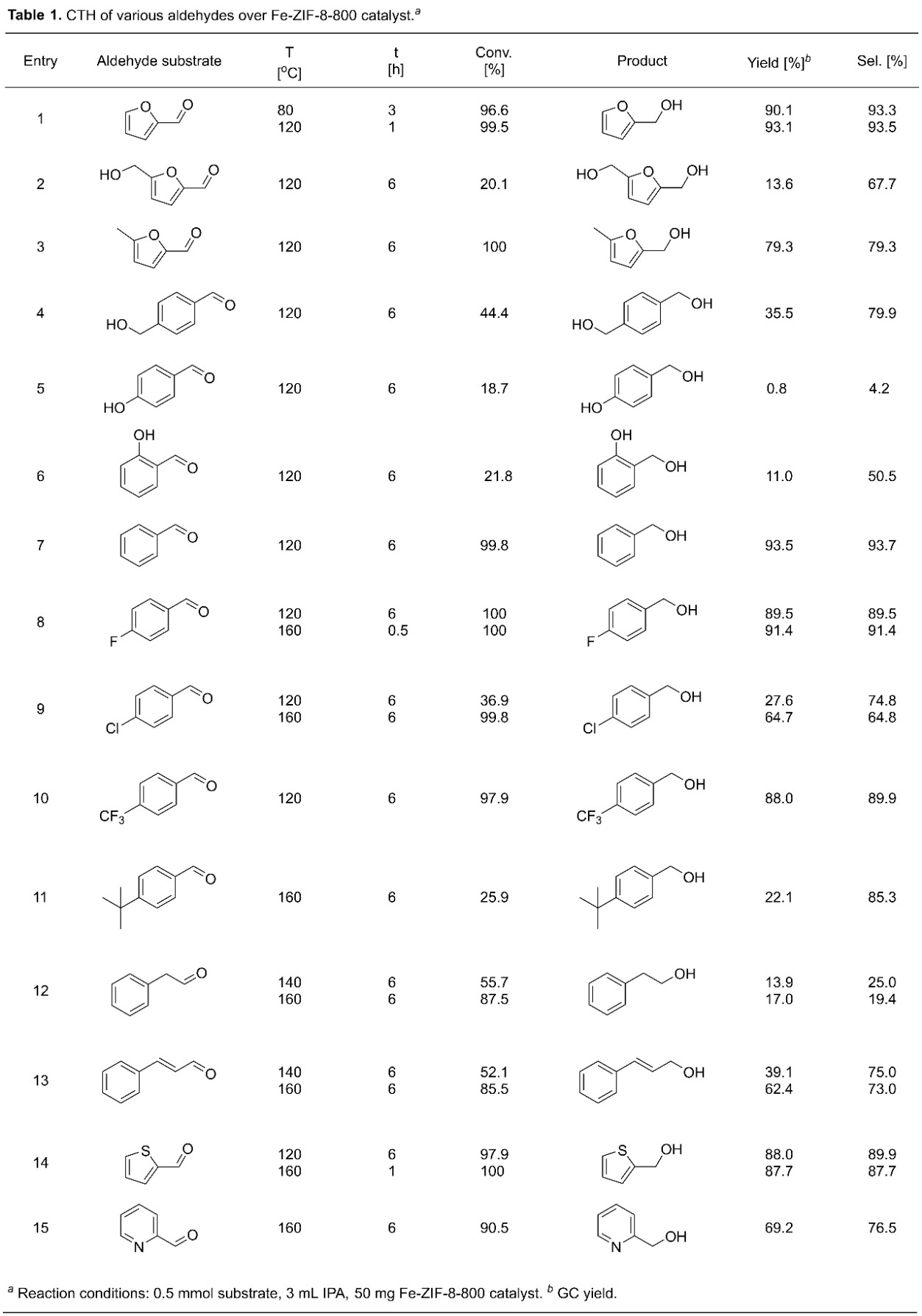
Finally, the effect of the substrate is investigated to demonstrate the versatility of the Fe-ZIF-8-800 catalyst (Table 1). HMF with an additional –CH2OH group gives poor yield of hydrogenated products and low conversion (entry 2). In contrast, a –CH3 on the ring gives a similar conversion but a slightly lower yield to the hydrogenated product (entry 3). The reactivities of aromatic molecules with a phenolic or primary –OH group and a –CHO group (entries 4 and 5) are again inferior. The conversions of p-hydroxybenzaldehyde and p-(hydroxymethyl) benzaldehyde is 18.7% and 44.4%, respectively, affording hydrogenated products with yields of 0.8% and 35.5%, respectively. In contrast, 93.5% yield to benzyl alcohol at 99.8% conversion of benzaldehyde is obtained at identical reaction conditions (entry 7).
In prior work, some of us have seen an electronic effect (either electron withdrawal or donation) of the substituent of the ring on the chemistry [55]. In addition, steric effects, competitive adsorption, and, in general, conformational effects, e.g. [56, 57], in adsorption of the substrate arising from different side groups can be at play. Finally, interactions of the functional groups of the ring with the solvent, e.g. [58], can significantly modify the adsorption of the substrate and/or the structure of the solvent around the active site and its ability to carry out the MPV reaction. Entries 5–9 in Table 1 indicate that electronic effects are probably not dominant here. In contrast, the additional –OH group in the substrate (entries 5 and 6) has a significant effect on conversion. The acidity of H atoms in primary and phenolic –OH groups is stronger than in IPA (H donor), and the CTH yields are related to pKa of the H atoms (Fig. 4). These acidic H atoms interact with the active centers of Fe SAs, resulting in inferior CTH reactivities, consistent with the differences in binding energies of –OH groups in HMF and IPA and the adsorption geometries (Figure S20).
The substitution of the p-position of benzaldehyde with –F and –CF3 groups leads to slightly lower yields of hydrogenated products, while –Cl leads to much lower selectivity (entries 8–10). The CTH reaction is greatly suppressed with p-substituted t-butyl group, probably due to the steric effect, inhibiting the adsorption of the substrate (entry 11). The reactivity of 2-phenylacetaldehyde is much lower, indicating that the activity of such –CHO group is lower than the benzylic –CHO group (entry 12). In comparison, an unsaturated aldehyde, such as cinnamaldehyde, exhibits better reactivity (entry 13). The replacement of the furan ring with a thiophene ring leads to a slightly lower yield of hydrogenated products, while the pyridine ring results in a much lower product yield (entries 14 and 15). Overall, the catalyst exhibits good chemoselectivity towards CTH of –CHO group without ring hydrogenation but its effectiveness is, as expected, substrate dependent.
Catalyst Recyclability
Recyclability experiments of the Fe-ZIF-8-800 catalyst (Figure S21a) show that the CTH activity gradually decreases after the 3rd run, whereas the selectivity to FA is nearly unchanged. The XANES and EXAFS spectra of used Fe-ZIF-8-800 catalyst (Figure S22) show that the Fe-N coordination number in the spent catalyst is slightly higher than that of the fresh Fe-ZIF-8-800 (4.56 vs. 3.7) (Table S8), while the Zn-N coordination is less changed (4.07 vs. 3.89) (Table S9). We conclude that the Fe-N coordination structures greatly influence the catalytic activity of the Fe catalysts. The activity of the spent catalyst can easily be recovered through calcination with additional trace Fe precursor, further confirming that trace Fe is indispensable for the excellent CTH performance of Fe-ZIF-8-800.
{kind=link}