A random colonization of human gut microbiome into conventional mice through FMT had a different impact on muscle strength.
The ongoing debate about the regulation of muscle strength by intestinal bacteria prompted us to explore a new approach to identify the specific bacteria responsible. Utilizing the concept of subgroup analysis, which helps uncover the cause of complex issues in large data sets, we performed a novel in vivo experiment. The aim was to identify the intestinal bacteria that regulate muscle strength through randomized subgroup analysis. We achieved this by feeding a fresh fecal sample to conventional mice (C57BL/6) whose gut microbiome had been depleted through a mixture of 3 broad-spectrum antibiotics and nystatin (as shown in Fig. 1a).
The effect of the subset FMT with the human gut microbiome in conventional mice was vastly different on muscle strength, as shown in Fig. 1b,c. In order to exclude genetic factors and evaluate the effect of the gut microbiome on muscle strength change alone, the rotarod records were monitored in the same mice after replacing their original gut microbiome with human gut microbiome through the FMT. The changes in the muscle strength of the mice over the three-month experimental period can be grouped into three categories: the group with increased muscle strength, in which the holding time on the rotarod increased by 23.5 ± 5.7 seconds (SMS); the group with unchanged muscle strength, in which the holding time on the rotarod remained within the range of 1.6 ± 0.8 seconds (MMS); and the group with decreased muscle strength, in which the holding time on the rotarod decreased by -16.1 ± 3.2 seconds (WMS) (Fig. 1b,c).The changes of muscle strength in each group were confirmed by the histological examination, which showed a high degree of muscle fiber accumulation in SMS, medium accumulation in MMS, and the lowest accumulation in WMS (Fig. 1d). The levels of blood glucose and lipid profiles did not change significantly before or after the gut microbiome replacement in the mice (Figure S1). These results suggest that a different subset of the human gut microbiome randomly replaced the original gut microbiome in each mouse, resulting in different effects on muscle strength.
Different types of gut microbiome were established in each of the experimental mice after the FMT with human feces.
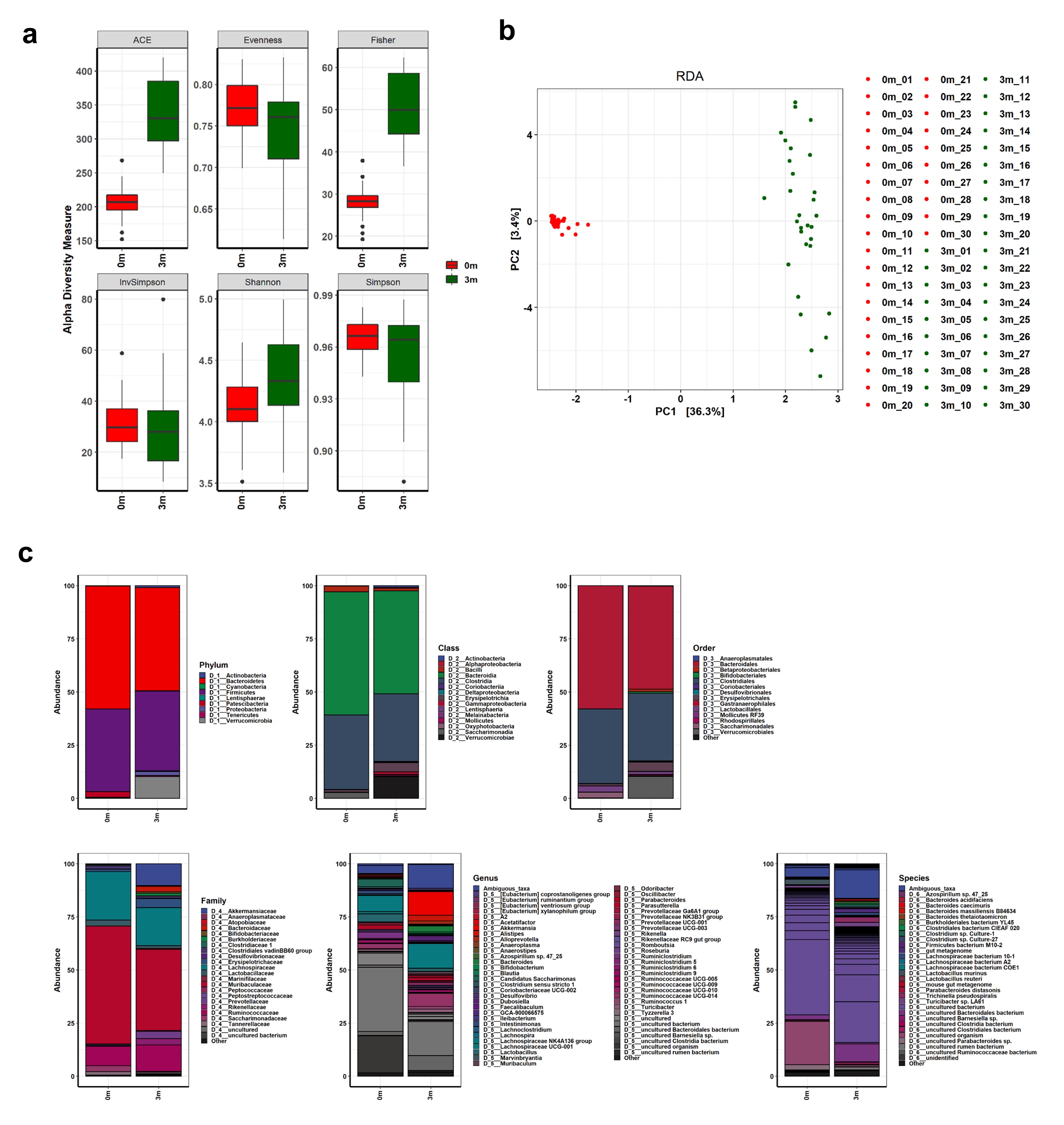
The differential effects on muscle strength after gut microbiome replacement prompted us to compare the compositional changes of the gut microbiome before and after FMT with human feces in the mice. We sequenced the V3-V4 regions of the 16S rRNA genes of the gut microbiome of each mouse before and after replacement using the MiSeq platform (Illumina). We filtered out the sequence reads that potentially contained incorrect primer or barcode sequences, sequences with more than one ambiguous base, low-quality sequences, or chimeras, which comprised about 0.001% of the total reads. The filtered 16S rRNA sequences were used to identify individual microbes by matching them to the SILVA reference database (region V3-V4) (https://www.arb-silva.de/). The identified 16S rRNA sequences, represented as operational taxonomic units (OTUs), were classified into nine different phyla: Bacteroidetes (48.471%), Firmicutes (37.456%), Verrucomicrobia (10.197%), Proteobacteria (1.924%), Patescibacteria (0.337%), Actinobacteria (0.907%), Tenericutes (0.48%), Cyanobacteria (0.228%), and Lentisphaerae (0.002%) (Figure S2 and Table S2). The OTUs were further classified to the species level.
The comparison of the OTUs showed a clear difference before and after the FMT in all of the mice, indicating that the replacement of their original gut microbiome with those of the humans was successful as shown in Figure S2. The differences were most notable at the genus and species levels (Figure S2c). In accordance with the individual variation in muscle strength after the FMT, the composition of the replaced gut microbiome was very different from one mouse to another (as shown in Fig. 2, Figure S3, and Table S2).
The differences in muscle strength were correlated with the shifts in the composition of the gut microbiome.
After confirming the individual differences in the composition of the replaced gut microbiome and its correlation with muscle strength, the gut microbiome of each mouse was analysed. The α-diversity metrics, which measure both richness and evenness, showed that different subsets of the human gut microbiome were replaced in each group of mice (as shown in Fig. 3 and Table S3). Although the α-diversity indices considering both richness and evenness (Shannon, Simpson, and InvSimpson) showed slight differences between the three groups, separate measurements of richness and evenness visualized the structural differences in the ecological community. The evenness indices before and after the FMT were similar, but the richness indices were increased after the FMT, suggesting that the human gut microbiome is more diverse, although the compositional characteristics of the human and mouse gut microbiomes are similar. Furthermore, the richness and evenness diversity indices of the mice with stronger muscle strength (SMS) were higher than in the other two groups, indicating a more diverse gut microbiome in SMS.
Other than α-diversity analyses, β-diversity analyses have also confirmed that the replaced gut microbiome is much more diverse than its original gut microbiome in each mouse. As shown in Fig. 4a,b, both β-diversity metrics, as measured by NMDS and MDS plots, show that the features are more diverse after the gut microbiome replacement.
To examine the impact of gut microbiome on muscle strength, we compared the gut microbiome composition before the FMT (T0) and three months after the FMT (T3). The comparison using unsupervised hierarchical clustering of the most abundant Operational Taxonomic Units (OTUs) based on the Bray Curtis distance revealed that the replacement of the gut microbiome affected the three groups of mice differently. The unsupervised hierarchical cluster analysis at T0 showed that each group of mice was completely distinct from each other (as seen in Fig. 4c, left). However, when considering muscle strength differences, individual mice from the three groups clustered together (as seen in Fig. 4c, right). This study only took into account muscle strength changes within individual mice, so the gut microbiome at the starting point did not show any group differences because the starting point was not related. However, the gut microbiome's impact on muscle strength is evident in the unsupervised hierarchical cluster analysis at T3, which shows the gut microbiome composition grouping individual mice according to differences in muscle strength.
Different microbial communities were established in each of the three groups of mice following the FMT.
The differences between the gut microbiomes of the three groups of mice (SMS, MMS, and WMS) were analysed by constructing phylogenetic trees. A maximum-likelihood phylogeny of each microbiome was built based on the 16S rDNA sequence (Fig. 5). The diversity of the gut microbiomes of all three groups expanded after the replacement of the gut microbiome: 66.2% in SMS, 46% in MMS, and 62% in WMS at the species level (Fig. 5). In addition to the increased diversity, the composition of the intestinal bacteria in the gut microbiome changed dramatically after the replacement.
Before the FMT, the main bacteria that constituted the original gut microbiomes at the phylum level were Bacteroidetes (56.093% in SMS, 53.922% in MMS, and 63.633% in WMS), Firmicutes (40.116% in SMS, 42.569% in MMS, and 33.938% in WMS), and Patescibacteria (3.24% in SMS, 2.791% in MMS, and 2.03% in WMS); at the class level, Bacteroidia (56.093% in SMS, 53.922% in MMS, and 63.633% in WMS), Clostridia (36.684% in SMS, 38.674% in MMS, and 29.873% in WMS), and Bacilli (2.489% in SMS, 2.499% in MMS, and 3.37% in WMS); at the order level, Bacteroidales (56.093% in SMS, 53.92% in MMS, and 63.628% in WMS), Clostridiales (36.684% in SMS, 38.674% in MMS, and 29.873% in WMS), and Lactobacillales (2.489% in SMS, 2.499% in MMS, and 3.37% in WMS); and at the family level, Muribaculaceae (54.598% in SMS, 50.66% in MMS, and 61.67% in WMS), Lachnospiraceae (24.471% in SMS, 24.611% in MMS, and 19.667% in WMS), and Ruminococcaceae (9.72% in SMS, 10.737% in MMS, and 7.399% in WMS). However, the composition of bacteria changed three months after the FMT: at the phylum level, Bacteroidetes (52.068% in SMS, 43.928% in MMS, and 49.416% in WMS), Firmicutes (36.244% in SMS, 37.628% in MMS, and 38.496% in WMS), and Verrucomicrobia (7.552% in SMS, 14.915% in MMS, and 8.123% in WMS); at the class level, Bacteroidia (52.068% in SMS, 43.928% in MMS, and 49.416% in WMS), Clostridia (30.437% in SMS, 32.948% in MMS, and 32. 293% in WMS), and Verrucomicrobiae (7.552% in SMS, 14.915% in MMS, and 8.123% in WMS); at the order level, Bacteroidales (56.093% in SMS, 53.92% in MMS, and 63.628% in WMS), Clostridiales (36.684% in SMS, 38.674% in MMS, and 29.873% in WMS), and Lactobacillales (2.489% in SMS, 2.499% in MMS, and 3.37% in WMS); at a family level, Muribaculaceae (40.613% in SMS, 33.303% in MMS, and 40.784% in WMS), Lachnospiraceae (16.383% in SMS, 18.101% in MMS, and 19.08% in WMS), and Ruminococcaceae (12.561% in SMS, 13.346% in MMS, and 11.583% in WMS) (Table S2). These results indicate that the human gut microbiome is not only more diverse, but also its composition significantly differs from that of mice.
The phylogenetic analysis showed that the replaced gut microbiomes of SMS, MMS, and WMS were different, despite the fact that their original gut microbiomes were not different from each other (Fig. 5). Consistent with the phylogenetic analysis, the co-occurrence network analysis also demonstrated that the microbial communities became more diverse after the gut microbiome was replaced in all three classified groups (Fig. 6 and Table S4). The 21 communities in SMS before the gut microbiome replacement expanded to 74, while the 28 communities in MMS became 60 and the 31 communities in WMS became 58. There was no significant difference in the number of nodes and edges within the microbial communities, which suggests that bacteria interact with each other in a similar manner. The higher number of microbial communities in SMS compared to MMS and WMS indicates that muscle strength is associated with a more diverse gut microbiome.
Intestinal microbes that affect muscle strength were identified at the species level.
The above gross gut microbiome analyses revealed a clear correlation between gut microbiome composition and muscle strength. However, the group analyses did not visualize a specific group of bacteria responsible for either promoting or reducing muscle strength (Figs. 2–6). Due to the limitations of group analysis in identifying intestinal microbes at the species level, we utilized the concept of fold change at the log2 scale and a linear correlation to analyse the abundance of intestinal microbes in relation to muscle strength (Fig. 7). Among the bacterial species, Phocaeicola barnesiae, Eisenbergiella massiliensis, and Anaeroplasma abactoclasticum were most abundant in SMS, indicating a positive correlation with muscle strength. On the other hand, Ileibacterium valens and Ethanoligenens harbinense were most abundant in WMS, indicating a negative correlation with muscle strength. The bacteria associated with strong muscle strength were classified under the phylum Bacteroidetes, Firmicutes, and Tenericutes, while those associated with weak muscle strength belonged to the phylum Firmicutes.
{kind=link}
{kind=link}
{kind=link}