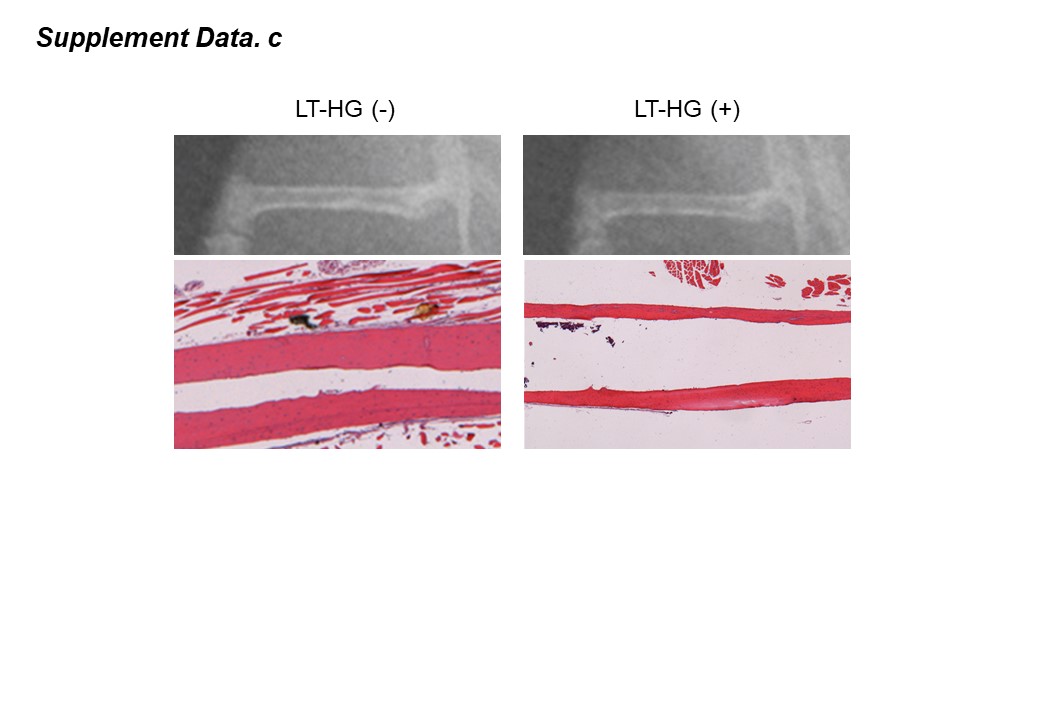
The main feature of this study was that a LT-HG model was created and used. As compared to the study by Kasahara et al. that used a short-term hyperglycemia model, the LT-HG model showed significant thinning of the femoral cortex (Supplemental Data. c) and increased BM adiposity and bone fragility against blunt external force [13]. These results better approximate real DM clinical conditions.
It has been widely reported that bone formation reduces under hyperglycemia. Recently, it has been reported that the proliferative capacity of human BMCs is reduced after 2 weeks of culture under hyperglycemia [15, 16] and that induction of differentiation of mouse BMCs under hyperglycemia promotes apoptosis of BMCs [10]. In addition, some studies have reported that the differentiation of BMCs into adipocytes is promoted by hyperglycemia and that differentiation into osteoblasts is impaired. Inhibition of osteoblast maturation in cultured mouse tissue [7, 8], impaired osteoblast differentiation, and reduced mineralization in mouse BMCs have also been reported [10]. However, these reports are from in vitro models in which tissue or cells themselves are cultured under hyperglycemia, and there are no reports of in vivo studies in which LT-HG fracture models were created and studied.
Regarding bone resorption capacity under hyperglycemia, it has been reported that there is no difference in osteoclast function between hyperglycemic and nonhyperglycemic mice because there are no significant differences in bone metabolism markers (serum TRAP5b, urinary DPD) or proteolytic enzymes (cathepsin K) [7, 8]. Conversely, in bone calluses of mice treated with STZ injection and hyperglycemic for 3 weeks, osteoclast function is reportedly enhanced due to an increase in TRACP cells, aggrecanases (ADAMTS), and collagenases (MMP-13,16) [17]. In a human control study, type 2 DM patients have been reported to have lower bone resorption capacity due to lower bone metabolism markers (serum TRAP5b, serum CTX) [18]. As described above, there is no consensus regarding osteoclast function and bone resorption capacity under hyperglycemic conditions, so findings from LT-HG models, like the one in the present study, are considered important. Based on research by Kasahara et al., we believe that osteoclast function is reduced [13].
The callus in the LT-HG model of this study was characterized by delayed chondrocyte resorption, decreased osteoblast aggregation, impaired multinucleation of osteoclasts, and narrowing of neovascular vessels. This study also found that the arrangement of osteoblasts became irregular, the gap between cells was large, and the number of aggregated cells tended to be small. These results, similar to previous experiments with cultured cells, suggest that hyperglycemia exposure impairs BMCs, resulting in impaired differentiation and maturation into osteoblasts and abnormal osteoblast morphology. This study compared results at 2–3 weeks after fracture, as Kasahara et al. found the greatest difference in callus characterization during this time [13]. As a result, we freshly observed that osteoclasts in the callus did not increase in cell number and had fewer nuclei when LT-HG was maintained. This clearly indicates that BMCs are impaired under LT-HG exposure and that bone resorption capacity is reduced due to impaired differentiation and function of osteoclasts and osteoclasts. The results of this study suggest that the bone metabolic capacity of the LT-HG model has decreased function in both bone formation and bone resorption.
Insulin producing cells normally occur only in the pancreas and thymus [19]. However, Kojima et al. reported that hyperglycemia in mice and rats induces the expression of proinsulin positive cells, an insulin transcript, in the liver, adipose tissue, and bone marrow, and that hyperglycemia induces the appearance of proinsulin-producing cells in the BM [20]. Terashima et al. reported that diabetic neuropathy in mice is caused by cell fusion of proinsulin-positive cells in the sciatic nerve and dorsal root ganglia, with the fused cells expressing TNF-α and undergoing a high rate of apoptosis [12]. Diabetic neuropathy in STZ-treated DM mice is also reported to require proinsulin-positive BMCs to produce TNF-α [21]. This result suggests that proinsulin-positive cells appear at various sites. In this study, the expression of proinsulin and TNF-α was also observed in the callus area in the LT-HG model, suggesting that the BMCs themselves may be damaged by the same mechanism as diabetic neuropathy, resulting in a prolonged fracture healing process. These results suggest that proinsulin may be a potential biomarker for the early diagnosis of diabetic tissue damage.
Regarding the role of TNF-α, there are reports from BMT experiments in DM mice that diabetic BMCs themselves may express TNF-α and cause glomerular damage [22] and that diabetic BMCs fuse with hepatocytes, resulting in TNF-α production and causing diabetic hepatocyte injury [23]. Diabetes-enhanced TNF-α also reduces MSC proliferation, increases MSC apoptosis, and inhibits osteoblast differentiation via Indian Hedgehog signaling [24, 25]. In this study, the increase in proinsulin-positive and TNF-α-positive cells in bone marrow derived cells (BMDCs) around small blood vessels in the callus are thought to impair the proliferation of MSCs and their differentiation into osteoblasts, thereby reducing their osteogenic potential.
Induction of MSCs from a local site such as the periosteum or BM is important for fracture healing. In addition, one of the important roles of MSCs is their involvement in hematopoietic stem cell (HSC) proliferation and maintenance of HSC progenitors [26, 27]. Since the callus at 2W post-fracture in this study was composed of many GFP-positive BMDCs, the induction of BMDCs into the matrix with angiogenesis is considered important in the fracture healing process. In addition, there were more proinsulin-positive and TNF-α-positive BMDCs in the LT-HG+ group, suggesting that LT-HG exposure may cause abnormalities in the BMDCs themselves.
One limitation of this study is the biological response such as cytokine release during fracture creation. To minimize the effects of fracture as much as possible, an osteotomy model was used instead of a blunt fracture model, in addition to soft tissue protection. The present study was limited to the second and third weeks after the fracture. However, in the study by Kasahara et al. [13], the greatest difference in callus formation was observed 2–3 weeks after fracture. In addition, Kayal et al. reported markedly increased expression of RANKL, TRACP, and TNF-α at 12 and 16 days after fracture in DM mice [17]. Therefore, the evaluation period for this study was judged to be sufficient. Future studies, however, may wish to address different time points after fracture.
{kind=link}
{kind=link}
{kind=link}