Ethics
The study protocol was approved by the local ethics research committee of Waseda University (Ethics Review Procedures Concerning Research with Human Participants; application number: 2021 − 423; approved on February 7, 2022). All procedures were conducted according to the ethics committee’s guidelines and regulations. All participants provided written informed consent before participating in the study.
Nukadoko maintenance and sample collection
The study participants were healthy volunteers recruited from acquaintances (N = 3); all were Japanese nationals, of which two were female, and one was male. The study was conducted in Tokyo, Japan, in February and March, 2022. The participants were given commercially available Nukadoko at the late stage and were asked to stir it for 14 days and not stir it for the remaining 14 days to examine microbial settlement and shedding on the skin. Nukadoko samples were collected on days 0, 3, 6, 9, 12, and 14 using individually wrapped disposable plastic spoons. Skin microbiome samples were collected on days 0, 3, 6, 9, 12, 14, 15, 18, 21, 24, 27, and 29 by swabbing the palm for 3 min using a sterile cotton-tipped swab (ESwab ™; Copan Diagnostics, Brescia, Italy). Swabs were stored in tubes with Liquid Amies Medium solution. (Copan Diagnostics, Brescia, Italy). Both sample types were immediately frozen and stored at -20°C until DNA extraction. The study workflow is illustrated in Fig. 2. The sampling duration for each Nukadoko and skin microbiome sample was at least 6 h.
Total DNA extraction and high-throughput sequencing
Samples were treated with 750 µL of lysis buffer from the GenFind V2 DNA extraction kit (Beckman Coulter, Indianapolis, IN, USA). The suspension was vortexed for 10 min, heat-treated at 100°C for 10 min, and centrifuged for 5 min at 20000 g. The supernatant was then mixed with EZ beads (AMR, Tokyo, Japan), and DNA was fragmented using the MM-400 unit (Retsch, Haan, Germany) at a maximum speed for 3 min. The remaining DNA purification steps were performed using the abovementioned GenFind V2 DNA extraction kit (Beckman Coulter), according to the manufacturer's protocol. DNA was eluted with 80 µL of nuclease-free water; using the KAPA HiFi HotStart ReadyMix (Roche, Basel, Switzerland)12,13 and specific primers (341F: 5'-TCGTCGGCAGCGTCAGATGTGTATAAGAGAGACACCTACGGGNGGCWGCA G-3') and 806R (5'-GTCTCGTGGGCTCGGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCT AATCC-3'), the V3–V4 region of the 16S rRNA gene was amplified. The thermal conditions were 95°C for 3 min, followed by 32 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 5 min. DNA samples, library preparation, and amplicon sequencing were performed using 300-bp paired-end sequencing on the Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA) at GenomeRead Inc. (Kagawa, Japan).
Microbiome analysis
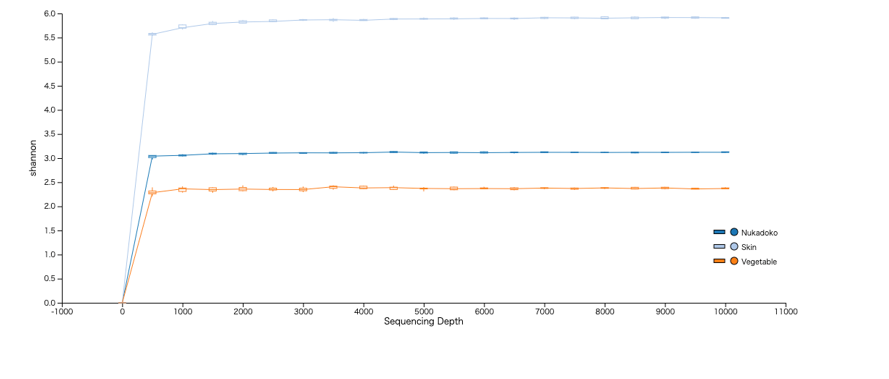
Microbiome analysis was performed as previously reported14. Briefly, raw FASTQ files were imported into the QIIME2 platform (2022.8) as qza files15. Denoising and read quality control were performed using the QIIME dada2 denoise-paired function, and reads were classified into ASVs16. We used 269 nt for-p-trunc-len-f and 255 nt for-p-trunc-len-r. The SILVA database’s SSU 138 (https://www.arb-silva.de/documentation/release-138/) was used with the QIIME feature-classifier classification scikit-learn package for taxonomic assignment17,18. ASVs classified as chloroplast, mitochondria, or unassigned were excluded from subsequent statistical analysis. Subsampling is a common method for inferring microbiome differences between samples and is a suitable analytical approach for analyzing new datasets. To evaluate the effect of sequence read counts on microbiome diversity assessment, we plotted changes in the Shannon index over a range of read counts from 0 to 10,000, using rarefaction curves. Subsampling was performed with a threshold of 10,000 reads (Supplementary Fig. 1).
Custom database for taxonomic assignment
The classifier database used in this study was made from Silva release 138.1 SSU 99% (www.arb-silva)17. Database curation was performed using REference Sequence annotation and CuRatIon Pipeline (RESCRIPt) following the developers’ recommended parameters19. Briefly, RESCRIPt removed low-quality sequences (sequences containing > 5 or more ambiguous bases or homopolymers of ≥ 8 bases) and filtered lengths (archaea [16S rRNA] ≥ 900 bp, bacteria [16S rRNA] ≥ 1200 bp, and eukaryotes [18S rRNA] ≥ 1400 bp). Additionally, deduplication of reads was performed in the Uniq mode. We then created the scikit-learn naive Bayes classifier using the QIIME2 plugin (feature classifier)18.
Calculation of shared ASVs
We defined shared ASVs as ASVs shared by > 1% of both datasets (Nukadoko and skin). When Nukadoko was touched for the first two weeks, data from Nukadoko and skin from the same day were used as pairs; when the bran was not touched for the next two weeks, data from Nukadoko from the last day and each skin microbiome data were used as pairs. The calculation was conducted using R (version 4.2.1) and phyloseq (version 1.40.0)20 or the custom python code (q2-shared_asv v0.2.0) with 0.01 for --p-percentage.
Principal component analysis
Principal component analysis (PCA) was performed using DEICODE—a tool that computes the Aitchison distance using tables of table data from QIIME221. DEICODE’s default parameters on the QIIME2 platform were used. Data were visualized using ggplot (version 3.4.0) and ggprism (version 1.40.0)22,23.
{kind=link}