Human embryonic stem cells derived retinal organoids faithfully recapitulate the initial neurogenesis of the human neural retina
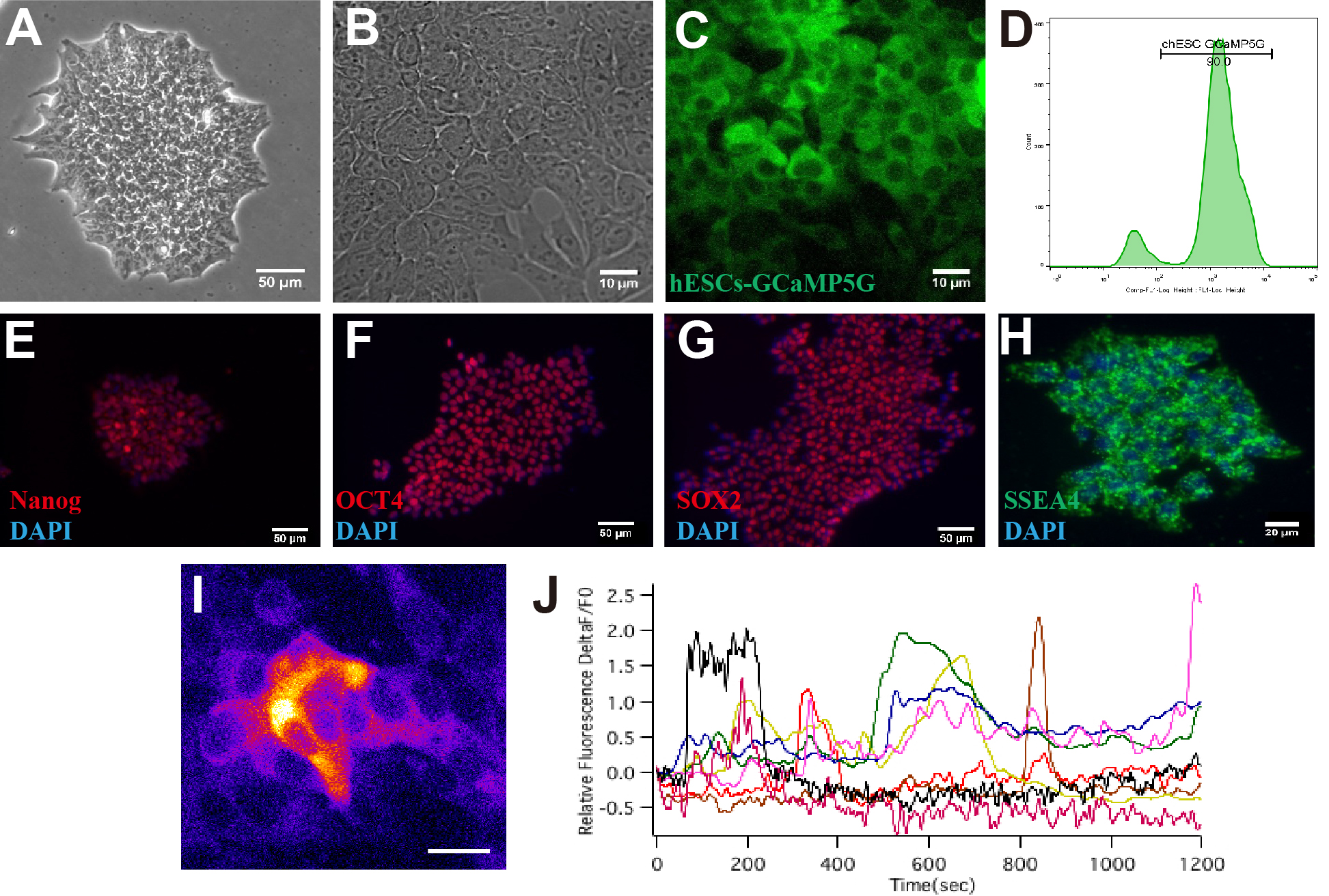
As the high consistency of early development between hROs and corresponding human fetal retina in vivo [17], to recapitulate the initial neurogenesis of human embryonic neural retina around six-week gestation, we determined to explore hROs from D24 to D30 when retinal ganglion cells were just born using a neural retina enriched induction protocol previously provided by Kuwahara et al [18]. Embryonic bodies acquired Rax+ optic vesicle with unlaminated epithelium morphology at D20 (Fig. 1A, B) and further developed into SOX2+CHX10+ neural retina at D24 (Fig. 1C). The hROs at D24 contain a large number of Math5+ retinal progenitor cells and differentiating p27kip1+ postmitotic cells at the basal layer of the neural retina (Fig. 1D, E). Pax6+ strong retinal ganglion precursor cells first emerged at Sox2− basal layer, accompanying the primitive lamination of neural retina at D24 (Fig. 1F). Retinal ganglion precursor cells continuously differentiated into Islet1+Tuj1+ immature RGCs at D30 (Fig. 1G). The immature RGCs turned to a mature phenotype with extending outward Tuj1 + axonal processes after hROs were attached to the gelatin-coated plate (Fig. 1H, I). To detect the molecular dynamic of the initial neurogenesis of neural retina in hROs, we constructed a human embryonic stem cell line expressing GCaMP5G, a genetically encoded calcium indicator suit for neural activity imaging [19], with pLOV-CMV-GCaMP5G transfection. More than 90% of cells expressed GCaMP5G indicating differential cellular calcium dynamic in human embryonic stem cell clones and sustained standard cell morphology and pluripotency after flow cytometry sorting (Additional fle 1:Fig.S1; Additional fle 3:Video1). After induction for 24 days, we again harvested hROs with GCaMP5G expressed in the neural retina (Fig. 1J). Retinal progenitor cells at the outer neuroblast layer were clearly labeled with GCaMP5G (Fig. 1K) and exhibited spontaneous robust global calcium transients which are representative of initial neurogenesis of neural retina [9] (Fig. 1L). These data supported that hROs from D24 to D30 well recapitulate the initial neurogenesis of the human neural retina around six-week gestation in vivo at the histological, cellular, and molecular level, distinguishing our work from previous model animal research.
Ethanol exposure slows the growth of the neural retina in hROs
Ethanol, a well-known teratogen for human embryonic development, has a role as an antiseptic drug, a polar solvent, a central nervous system depressant, and a disinfectant. Prenatal alcohol exposure during the initial neurogenesis of the neural retina is catastrophic for eye development. To address the impact of ethanol on the initial neurogenesis of the human neural retina, we first assessed the morphological change of hROs after ethanol exposure. The hROs were stochastically divided into four groups which treated ethanol from D24 to D30 with 0% (control, v/v), 1%, 2%, and 3% EtOH, respectively. As hROs continuously grow in the control condition, 3% EtOH directly brought hROs to a dehydrated-like morphology. Treatment with 2% EtOH induced hROs an atrophic phenotype, whereas 1% EtOH almost aborted the growth of neural retinal in hROs (Fig. 2A). The thickness of neural retina at D24 stayed at about 185 ± 23µm (mean ± SD), grew to 249 ± 24µm during the development. Treatment of 1% EtOH resulted in a suspension of thickness at 187 ± 26µm. In comparison, 2% EtOH resulted in a shrinkage of thickness at 142 ± 40µm (Fig. 2B). Calculation of variance of neural retina thickness also reflected a significant reduction of growth with 1% EtOH and a negative with 2% EtOH (Fig. 2D). In line with this, measurement of the maximum sectional area of hROs and its variance between D30 and D24 also indicated a similar trend that hROs slowly ceased to grow with 1% EtOH and atrophied with 2% EtOH (Fig. 2C, E). To find the most significant altered mechanism in the maximum pathophysiological condition of heavy drinking rather than the toxicology of ethanol on the eye development, we selected 1% EtOH as a concentration for further experiment, lower than that previously described in the zebrafish model research [20].
Ethanol exposure causes neuronal differentiation defect and cell death in neural retina of hROs
Previous studies have focused on the role of ethanol exposure on optic vesicle evagination at an far early stage [21] or the maturation of the optic nerve around the late pre-/postnatal stage [22]. As we found that 1%EtOH robustly reduced the growth rate of the neural retina at the initial neurogenesis stage, we sorted out to investigate the generation of retinal ganglion cells (RGCs), the first-born neuron in the neural retina. Compared with the control group, Pax6+ strong retinal ganglion precursor cells robustly decreased with 1% EtOH (Fig. 3A, B, I). Moreover, radial migration of immature Tuj1+ RGCs along the Nestin + scaffold was impaired, leading immature Tuj1 + RGCs failed to correctly locate at the basal layer of the neural retina (Fig. 3C, D). When attached to the gelatin-coated dish for a week, RGCs in the neural retina could extend their axons out of hROs for up to 2.0 mm long with dense spines in the control condition (Fig. 3E, G, K). However, 1% EtOH weakened the ability of axonal extension and spine maturation of RGCs and leads to a reduction in the axon density, axonal length, and spine density after hROs attachment (Fig. 3F, H, J ~ L).
As the cell death and cell cycle disruption accounts for most ethanol induced neurodevelopmental defects in previous animal model research [23]. Hence, we further investigated the cell death of hROs after ethanol treatment. Immunostaining showed the percentage of cleaved-caspase3+ cells in ethanol-treated hROs significantly elevated to more than 22.1 ± 7.6% (mean ± SD) compared with 4.9 ± 1.5% in control hROs (Fig. 4A ~ C), indicated a burst of regulated cell death after ethanol treatment. TUNEL assay showed a similar result: ethanol increased the percentage of TUNEL+ cells in hROs from 3.3 ± 0.7% to 23.8 ± 7.0% (Fig. 4D ~ F). To detect the cell cycle, we dissociated the hROs and analyzed the cell cycle with flow cytometric analysis. More than 59.6 ± 7.3% of cells stayed at G1 phase in the control condition while increasing to 82.5 ± 4.8% after ethanol treatment. Consistently, cells in S and G2/M phase dropped to 10.3 ± 2.5% and 7.2 ± 2.4% after ethanol treatment compared with the control group staying at 24.4 ± 5.4% and 16.0 ± 1.9%, respectively (Fig. 4G, H). This result indicated ethanol-induced cell-cycle arrest in the G1 phase of cells in hROs, which supports previous results from animal models [24].
Transcriptome profiling reveals the calcium signaling pathway as a candidate mechanism of cell death and neuronal differentiation defect in ethanol-treated hROs
To investigate the possible molecular mechanism beneath the ethanol-induced cell death and neuronal differentiation defect, we applied bulk RNA-seq analysis of both control hROs and ethanol-treated hROs. Bioinformatics analysis showed that there were a total of 434 (173 genes upregulated and 261 downregulated genes) differentially expressed genes (DEGs) between the control and ethanol-treated hROs (Fig. 5A). Cluster heat maps showed DEGs between the control hROs and ethanol-treated hROs (Fig. 5B). Volcano plots presented the distribution of DEGs as gray, red, and green circles (Fig. 5C). Enrichment analysis in the PaGenBase [25] demonstrated that these DEGs of ethanol-treated hROs could be enriched as retina tissue-specific pattern genes. Enrichment analysis in Cell Type Signatures [26] further identified DEGs of ethanol-treated hROs as embryonic neural stem cells of homo sapiens. To interpret these DEGs for specific biological processes, we next conducted the Gene Ontology (GO) annotation analysis. The terms such as chromosome organization, positive regulation of cell death, visual perception, regulation of cell development, regulation of neuron differentiation, regulation of ion transport, eye development, and response to calcium ion were significantly enriched for ethanol-treated hROs based on gene counts and P-value (Fig. 5F). This result aligned with the previous study about chromosome damage in ethanol-related diseases [27], and supported the biological phenomenon shown in Figs. 3 and 4. It is worth noting that ion transport and calcium ion response were also enriched as significant biological process. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation further distinguished several pathways such as systemic lupus erythematosus, amyotrophic lateral sclerosis, pathways in cancer, shigellosis, toxoplasmosis, circadian rhythm, and calcium signaling pathway (Fig. 5G). The chromosome related pathway and immune system disruption of teratogenic effects of ethanol exposure has been widely discovered, however the calcium signaling alteration induced by ethanol remained unknown. So, how ethanol altered the calcium signal pathway during initial neurogenesis of the human neural retina aroused our interest.
Ethanol exposure alters the dynamic of calcium signaling in hROs
The genetically encoded calcium indicator and two-photon microscope enabled us to capture the living cellular calcium signaling in hROs without chemotoxicity and phototoxicity. As shown, he neural retina of hROs presented multiple spontaneous calcium spiking regions characterized as global and local transients (Fig. 6A, B). These regions spontaneously actively generated calcium transients at 0.55 ± 0.46 events per minute (mean ± SD) in the local transient domain and at 0.46 ± 0.20 events per minute in global transient domain (Fig. 6C, O, R; Additional fle 3:Video 2). Amplitude and duration of the local calcium transient maintained at 8.5 ± 4.8 Z and 4.8 ± 4.3 seconds, respectively (Fig. 6D, M, N). Amplitude and duration of the global calcium transient maintained were at 4.0 ± 3.2 Z and 17.9 ± 15.8 seconds, respectively (Fig. 6E, P, Q). Ethanol exposure produced a profound effect on the number of calcium spiking domains contributed by a robust increase of local calcium transient (Fig. 6F, G, K, L). The spiking frequency of local and global transient reduced to 0.14 ± 0.08 and 0.16 ± 0.10 events per minute respectively after ethanol exposure (Fig. 6H, O, R; Additional fle 3:Video 3). For local calcium transient, ethanol exposure did not alter the amplitude but prolonged the duration to 16.2 ± 18.2 seconds (Fig. 6M, N). For global calcium transient, ethanol exposure reduced the amplitude to 2.3 ± 1.8 Z but did not alter the duration (Fig. 6P, Q). These data showed ethanol exposure could significantly increase the numbers of local transient domains, lower local calcium transients’ frequency, and prolong its duration. Moreover, ethanol exposure significantly lowered global calcium transients’ frequency and reduced amplitude. Together, these results supported that ethanol exposure altered the calcium signaling dynamic in hROs and may induce cell death and neuronal differentiation defect during the initial neurogenesis of the neural retina.
Identification of RYR1 and CACNA1S as regulators of calcium ion transport and RET as synergistic effecter in the calcium signaling pathway of ethanol-treated hROs
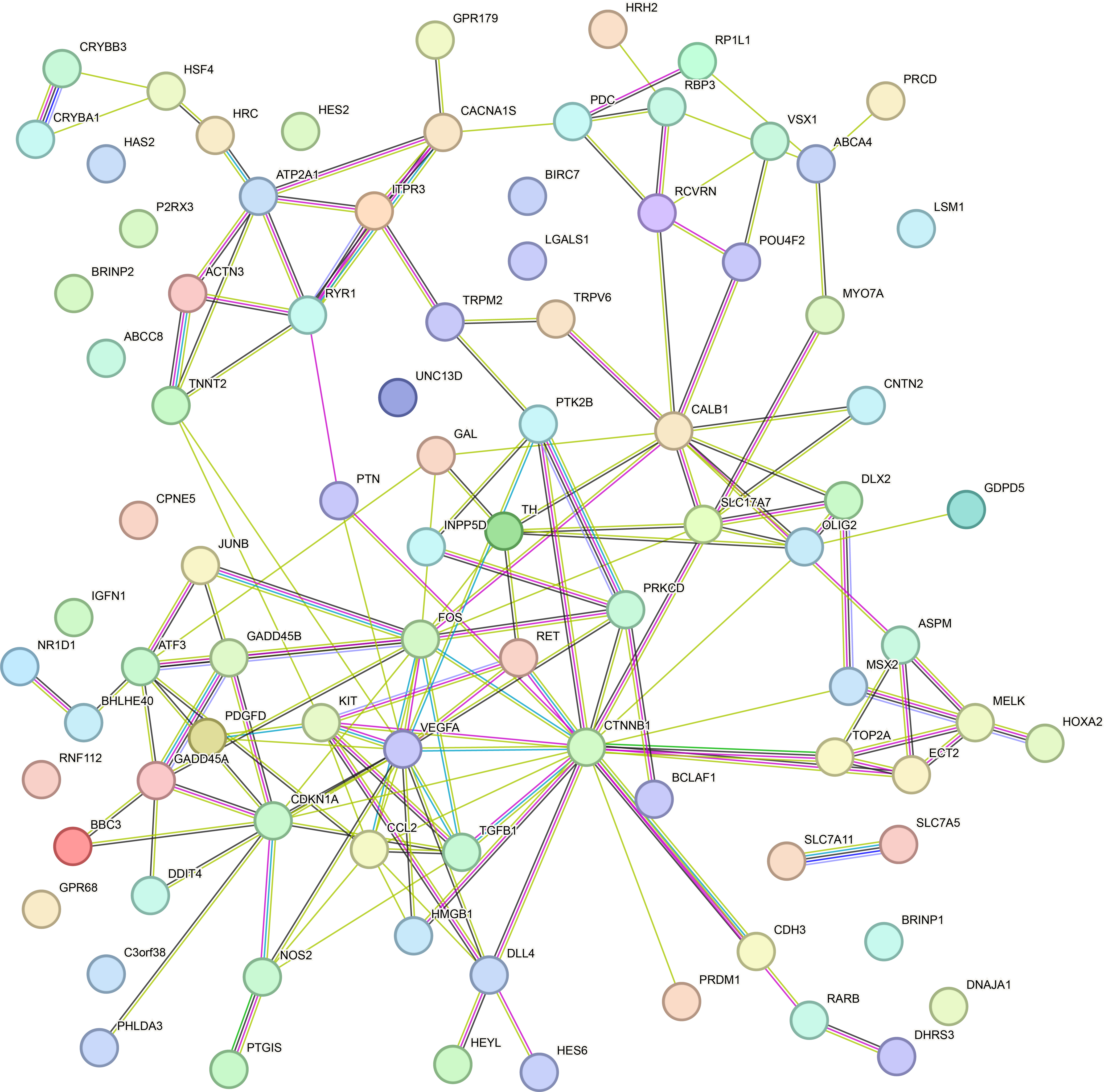
To uncover the potential molecular mechanism beneath the ethanol-induced calcium signaling alteration, we next performed the gene set enrichment analysis (GSEA) analysis of DEGs in ethanol-treated hROs to screen target genes of the calcium signaling pathway. As shown, genes involved in voltage-gated calcium channel activity were enriched, and the most significantly changed genes, such as RYR1 and CACNA1S, were selected as candidate target genes (Fig. 7B). RYR1 encodes the ryanodine receptor 1 on the endoplasmic reticulum and acts as a calcium channel to release the calcium into the cytoplasm [28]. CACNA1S encodes the α1S subunit of L-type voltage-dependent calcium channel expressed on the plasm membrane to transport extracellular calcium into the cytoplasm [29]. Both gene expressions were verified by qRT-PCR (Fig. 7C) and could help explain the calcium signaling alteration observed with the two-photon microscope. Besides, to investigate the effector gene of a downstream cascade of the calcium signaling pathway, we further performed Cnet plotting of their associated genes in DEGs of ethanol-treated hROs. In the network, calcium transporters, such as RYR1 and CACNA1S, were included, and the RET gene was highlighted as the most related target genes with only its degree ≥ 6 (Fig. 7A). RET has four cadherin-like domains binding of calcium ions and participating transduction of downstream effect of calcium signaling via MAPK- and AKT- cascade that plays a role in cell differentiation and survival. To further understand the critical nodes of the RET-related pathway in ethanol-treated hROs, we analyzed these DEGs with the STRING online database to predict the functional protein-protein interaction of RET (Additional fle 2:Fig.S2). Among these results, CTNNB1, VEGFA, KIT, and TH were predicted as candidate interactive proteins which has been demonstrated playing a role in the cell death/survival and neuron differentiation. As a member of the calcium signaling pathway, the Cnet plotting showed RET simultaneously participated communities, such as eye development, positive regulation of cell death, and regulation of neuron differentiation, and regulation of cell development. To verify this prospective relation, we confirmed mRNA levels of selected genes, such as the specific retinal ganglion cell transcription factor POU4F2,the anti-apoptotic gene BCL-2, and its transcriptional repressor, the pro-apoptotic gene BCLAF1, and RET. The result showed that the mRNA level of RET, BCL-2, and POU4F2 were downregulated, but BCLAF1 was upregulated in ethanol-treated hROs (Fig. 7C, D). This result indicated that RET acts as a center effector gene of a calcium signaling pathway in ethanol-induced cell death and neuron differentiation in hROs.
{kind=link}
{kind=link}