ROS positively regulated G6PD and NF-κB signaling pathway in ccRCC cells
A pilot study revealed that G6PD-facilitated ROS accumulation could positively induce G6PD transcription in RCC [19]. To further reveal the impact of ROS on G6PD expressional regulation and discover other relevant signaling factors involved in this process, ROS production was triggered or inhibited with H2O2 or NAC in ACHN, 786-O and Caki-1 cells. The results demonstrated that G6PD mRNA expression level was significantly decreased in NAC-treated ACHN, 786-O and Caki-1 cells, whereas it was obviously increased in H2O2-stimulated cells compared with the control (Fig. 1A-D). These results were consistent with previous study using different ccRCC cell lines [19], and further confirmed the reciprocal regulatory effect between ROS and G6PD in ccRCC. Therefore, the functional factors of signaling pathways simultaneously involved in ROS and G6PD regulation were analyzed using the MatInspector software platform (will be discussed later in Fig. 2A). The results indicated that the NF-κB signaling pathway, which was demonstrated to be dysregulated and functional in the ROS-related chemotherapeutic resistance of ccRCC cells [24, 25], might intervene in the regulation of G6PD expression. To verify this hypothesis, the protein expression pattern of several important functional factors of NF-κB pathway, including p105, p50, p65, pIκBα (S32+S36), and IκBα, which have been proved to participate in signaling transduction and closely link to ccRCC tumorigenesis [24, 25, 28, 41], was examined in NAC- or H2O2-treated ACHN, 786-O and Caki-1 cells. The results from Western blot showed that the levels of pIκBα, the most abundant inhibitory molecule of NF-κB [41], were reduced by ROS depletion, but there were increased following ROS accumulation in all these three cells. Moreover, ROS positively regulated both the NF-κB signaling activation and G6PD expression in ccRCC cells (Fig. 1B-D). Based on the previous conclusion that G6PD promoted RCC proliferation through positive feedback regulation of ROS-pSTAT3 axis [19], the present findings led to the hypothesis that ROS regulated G6PD overexpression might not only through pSTAT3 activation but also the NF-κB signaling pathway.
To further clarify the interactions between G6PD and NF-κB signaling pathway, TNFα or BAY11-7082, which could simulate or inhibit the activity of NF-κB signaling pathway, were used to treat 786-O and Caki-1 cells. Subsequently, the expression of G6PD and NF-κB pathway–related functional activators, including p50 and p65 at the protein level were evaluated. The results from western blot displayed that the protein expression levels of G6PD, p50 and p65 were increased in a dose-dependent manner in TNFα‑treated cells, whereas they were decreased in BAY11-7082‑treated cells. The phosphorylation of IκBα were increased or reduced following NF-κB signaling activation or inhibition, respectively (Fig. 1E-F). Additionally, the results from real-time RT-PCR demonstrated that the mRNA expression levels of G6PD was also increased in TNFα‑treated cells, whereas it was decreased in cells following BAY11-7082 treatment (Fig. 1G-H), which was consistent with the expression changes of G6PD at the protein level. Taken together, these findings suggested that ROS-mediated NF-κB signaling pathway may be involved in increased G6PD transcription in ccRCC.
NF-κB signaling pathway facilitated G6PD expression via direct protein–DNA interaction with p65 instead of p50
To explore the underlying mechanism of NF-κB signaling pathway regulated G6PD expression in ccRCC, MatInspector software platform was used to analyze the potential regulatory factors that could bind to G6PD promoter. Fortunately, the binding site of NF-κB signaling–related factor was found to occupy the –483 to –473 bp of the G6PD promoter (Fig. 2A). Subsequently, a sequence containing this potential binding site along with the confirmed pSTAT3 binding site on G6PD promoter [19] was cloned to pGL3-BASIC vector (named G6PD-luc), and luciferase report assay was performed to determine the impact of TNFα or BAY11-7082 on the luciferase activity in 293T cells. As presented in Fig. 2B-C, the luciferase activity of G6PD-luc was significantly increased by 4.2-fold following TNFα (100 ng/ml) treatment, whereas the luciferase activity was significantly decreased by about 37% following BAY11-7082 (5 μM) treatment. Additionally, repeated luciferase reporter assay using a mutant vector with the NF-κB-binding sequence deleted (G6PD-luc-D) displayed that the luciferase activity was significantly decreased compared with the wild type vector. Moreover, in TNFα-treated cells, the luciferase activity was increased by only 2.3‑fold; however, in BAY11-7082-treated cells, the luciferase activity was decreased by no more than 21% (Fig. 2D), which demonstrated that the regulatory effect of NF-κB signaling pathway on G6PD expression was obviously attenuated without NF-κB-binding sequence. These findings indicated that NF-κB signaling pathway may promote G6PD overexpression through a direct transcriptional regulatory effect.
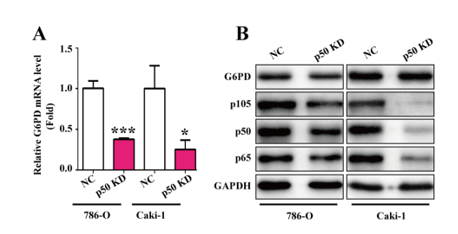
In order to explore how NF-κB signaling pathway contributes to G6PD transcription, oligonucleotide primers covering the potential NF-κB binding site were designed for ChIP analysis in ACHN, Caki-1, and 786-O cells by using anti-p65 or anti-p105/50 antibodies. The results demonstrated that p65 instead of p105/50 displayed a space-occupying effect (Fig. 2E), indicating that p65 might exert a direct regulatory effect on the G6PD promoter. To further evaluate the role of p65 in G6PD expression, the endogenous expression of p65 was knocked down with RNAi lentivirus in 786-O and Caki-1 cells. The results from real-time RT-PCR and Western blot demonstrated that both G6PD mRNA and protein expression levels were significantly declined compared with the negative control (Fig. 2F-G), which suggested that p65 could directly regulate G6PD transcription by occupying the binding site on the G6PD promoter. Additionally, the expression of G6PD mRNA levels was obviously decreased following p50 RNAi lentivirus transfection in 786-O and Caki-1 cells, whereas the results of Western blot analysis demonstrated that the G6PD protein expression levels were not significantly modified by p50 knocked down (Fig. 2F-G). Taken together, these findings confirmed the hypothesis that NF-κB signaling pathway facilitated G6PD expression via direct protein–DNA interaction with p65 instead of p50.
p65 and pSTAT3 presented a synergistic effect on G6PD transcriptional regulation
In a previous study conducted in our laboratory, it was found that G6PD was regulated by pSTAT3 through directly binding to –1614 bp to –1595 bp of the G6PD promoter region [19]. Interestingly, the potential p65-binding site is just adjacent to the functionally verified pSTAT3-binding site, which is located –184 to –166 bp on the G6PD promoter calculated from the transcriptional start site (Fig. 3A), indicating that pSTAT3 and p65 might co-occupy the binding region and interact with each other to synergistically promote G6PD overexpression. To testify this hypothesis, the ChIP assay was performed in three cell lines including ACHN, 786-O and Caki-1. Firstly, cell lysates were immunoprecipitated with anti-p65 or anti-p105/50 antibody, and the eluates were amplified with primers covering the pSTAT3-binding site (Fig. 3B). Conversely, cell lysates were immunoprecipitated with anti-pSTAT3 (S727) or anti-STAT3 antibody, and the eluates were amplified with primers covering the NF-κB-binding site (Fig. 3C). The analysis revealed that p65 instead of p50 was capable of binding to the pSTAT3-binding site (Fig. 3B), but neither pSTAT3 (S727) nor STAT3 interacted with the NF-κB effective binding site (Fig. 3C). These results indicated that pSTAT3 and p65 might form a complex and bind to the pSTAT3- rather than the NF-κB-binding site. In this G6PD transcriptional regulatory model, although p65 had the potential to regulate G6PD transcription independently, it could also form a p65/pSTAT3 transcriptional complex and occupy pSTAT3 but not its own binding site while displaying a synergistic effect with pSTAT3.
To consolidate this hypothesis, the Co-IP assay was conducted in ACHN, 786-O and Caki-1 cells to investigate the protein–protein interaction between pSTAT3 and p65. The results demonstrated that the substantial interaction between pSTAT3 and p65 indeed existed in ccRCC cells (Fig. 3D), which suggested that there may be an interaction between STAT3 and NF-κB signaling pathways on G6PD transcriptional regulation. Furthermore, results from luciferase report assay demonstrated that the luciferase activity of G6PD-luc containing both the NF-κB and pSTAT3 binding sites was significantly increased following the treatment of STAT3 or NF-κB signaling pathways activator (Fig. 3E), whereas it was decreased following the treatment of STAT3 or NF-κB signaling pathways inhibitor (Fig. 3F). Moreover, synergistic effects could be seen in combinative stimulation with these two signaling pathways activator or inhibitors (Fig. 3E-F). However, the luciferase activities were not obviously modified following NFκB and/or pSTAT3 signaling activator/inhibitors stimulation by using a mutant vector with both the NF-κB- and pSTAT3-binding sequence deletion (Fig. 3G-H). Taken together, these findings demonstrated that STAT3 and NF-κB signaling pathways presented a cross-talk and synergistic effect on G6PD transcription. The underlying mechanism was that p65 and pSTAT3 formed a p65/pSTAT3 complex, occupied the pSTAT3-binding site of the G6PD promoter, and synergistically facilitated G6PD overexpression in ccRCC.
NF-κB and STAT3 activated each other and facilitated ccRCC proliferation synergistically
The aforementioned results suggested that STAT3 and NF-κB signaling pathways over‑activation may synergistically contribute to ccRCC proliferation following G6PD overexpression. To further evaluate the reciprocal effect between these two pathways and the role of NF-κB/STAT3-mediated G6PD overexpression in ccRCC growth, 786-O cells were stimulated with the STAT3 signaling activator (IL-6) or inhibitor (STATTIC) to promote or inhibit STAT3 phosphorylation, respectively. The results from Western blot showed that the expression of p50, p65, and pIκBα were significantly increased following treatment with IL-6, whereas they were decreased following treatment with STATTIC (Fig. 4A), which suggested that the STAT3 phosphorylation status could influence the activation of NF-κB signaling pathway. Likewise, NF-κB over‑activation or inhibition could also positively impacted the STAT3 signaling pathway activation in a dose-dependent manner (Fig. 4B). These findings indicated a reciprocal regulatory effect between NF-κB and STAT3 signaling pathways in ccRCC.
It has been reported that aberrant NF-κB activation depends on CDK4/6-mediation and contributes to tumor cell progression [42]. Previous results from our laboratory demonstrated that pSTAT3 excessive activation contributed to G6PD-stimulated RCC cell proliferation via upregulated CyclinD1 expression [19]. In the present study, the role of NF-κB and STAT3 cross-talk in ccRCC growth was further explored. It was found that NF-κB positively regulated not only CyclinD1 but also CDK4 expression at the protein level (Fig. 4B), which suggested that NF-κB might synergistically cooperate with STAT3 and in part contribute to ccRCC proliferation following G6PD overexpression. To confirm this hypothesis, endogenous p65 expression was knocked down by infected 786-O and Caki-1 cells with RNAi lentivirus. The results from real-Time RT-PCR and Western blot demonstrated that the expression of STAT3, CyclinD1 and CDK4 were significantly decreased at the mRNA and protein level in p65 knocked down cells (Fig. 4C-E). These findings suggested that NF-κB and STAT3 signaling pathways could activate each other and exhibit a synergistical proliferation-promoting regulatory effect in ccRCC.
To further evaluate the role of NF-κB/STAT3-mediated G6PD overexpression in ccRCC growth, the MTS assay was conducted to determine the proliferation rate of ACHN and 786-O cells following treatment with STATTIC or BAY11-7082, respectively. As shown in Fig. 4F-G, both NF-κB and STAT3 inhibition presented a proliferation-inhibiting effect on ACHN and 786-O cells. However, whether G6PD overexpression is involved in this growth suppression effect needs to be clarified. Therefore, further MTS analyses were performed in G6PD‑overexpressing ACHN and the relevant control cells to detect the proliferation-inhibiting effect of STATTIC or BAY11-7082. As presented in Fig.4H-I, G6PD overexpression could reversed the impact of STAT3 and NF-κB inhibitor on ACHN cell proliferation. Furthermore, ACHN or 786-O cells were treated with STATTIC and BAY11-7082 independently or in combination. The results from MTS assay demonstrated that these two inhibitors displayed a synergistical growth suppression effect in both ACHN and 786-O cells (Fig. 4J), and the overexpression of G6PD in ACHN cells, which had the lowest G6PD activity in the three of the RCC cell lines, could rescue this tendency (Fig. 4K), indicating that G6PD overexpression is necessary for NF-κB/STAT3 promoted ccRCC proliferation. Taken together, these results suggested that NF-κB and STAT3 signaling pathways could activate each other, synergistically promote G6PD expression and contribute to ccRCC proliferation.
G6PD, pSTAT3, and p65 were highly expressed and positively correlated with each other in ccRCC tissues
To verify the aforementioned mechanistic model in vivo, G6PD expression profile was evaluated in 27 ccRCC tumor species and matched adjacent normal tissues by real-time RT-PCR. The results demonstrated that 16 of 27 (59.3%) tumor species exhibited high G6PD expression levels (Fig. 5A), and the statistical analysis revealed that the mRNA expression level of G6PD was significantly increased in ccRCC species compared with normal tissues (Fig. 5B). Subsequently, all the specimens were subjected to Western blot analysis for the protein expression level detection of G6PD, pSTAT3, and p65. The results demonstrated that the expression levels of all these factors were significantly increased in ccRCC tumor tissues compared with adjacent normal tissues (Fig. 5C-F). In addition, the results from Pearson correlation analysis demonstrated that G6PD, pSTAT3, and p65 were positively correlated with each other at the protein level (Fig. 5G-I). Moreover, immunohistochemistry staining was performed to analyze the expression of G6PD, pSTAT3, and p65 in tumor tissues and adjacent normal tissues. As shown in Fig. 5J, G6PD and pSTAT3 was mainly localized in the cytoplasm and nucleus of the ccRCC tumor cells, respectively; whereas the predominant localizations of p65 was seen within the whole ccRCC cells. Moreover, G6PD along with both pSTAT3 and p65 were displayed high expression levels in tumor tissues compared with adjacent normal tissues (Fig. 5J-K). Taken together, these findings demonstrated that the persistent activation of both NF-κB and STAT3 signals existed in ccRCC tissues, which may contribute to ccRCC tumorigenesis partially through the synergistically mediated G6PD overexpression.
G6PD activity inhibition attenuated the growth of ccRCC cells both in vitro and in vivo
The aforementioned results showed that NF-κB and pSTAT3 signaling pathways were likely to drive G6PD overexpression through the constitutively activated effect of p65 and pSTAT3, indicating that G6PD, as an important common mediator of NF-κB and pSTAT3 signaling, might become a potential therapeutic target for ccRCC treatment. Therefore, it was necessary to investigate whether G6PD activity inhibitors had an anti-tumor effect both in vitro and in vivo. The G6PD competitive activity inhibitor 6-aminonicotinamide (6-AN), a nicotinamide analog [43], was first tested in 786-O cells, one of the most highly cited cell lines that exactly clustered with ccRCC and commonly used for xenografts studies [39]. The results demonstrated that the G6PD activity was significantly decreased in a dose-depcendent manner in 786-O cells following treatment with 6-AN (Fig. 6A). Moreover, the anti-proliferative effect was seen in 786-O cells after treated with 6-AN at the indicated time points and doses (Fig. 6B). In addition, the 786-O parental tumor-bearing nude mice were treated intratumor with vehicle or 6-AN to evaluate the tumor-inhibitory activity of 6-AN in vivo. As presented in Fig. 6C-E, both the tumor volume (Fig. 6C-D) and tumor weight (Fig. 6E) were reduced following 6-AN treatment compared with the vehicle. Subsequently, the expression of G6PD, p-STAT3, and p65 in tumor xenografts was compared by Western blot analysis. The results showed that the expression levels of these factors were decreased in the 6-AN-treated tumor compared with the vehicle (Fig. 6F-G). Taken together, these in vivo data confirmed that there may be a positive correlation between G6PD overexpression and NF-κB/STAT3 signaling pathway over-activation. Moreover, G6PD inhibition exhibited tumor-suppressing activities in ccRCC, indicating that G6PD might be a potential therapeutic target for ccRCC treatment.
{kind=link}