Source of chemicals, antibodies, composition of buffers, equipment and software used in this study are listed in supplemental Table S1.
Recombinant AAV production and purification
Vector production and purification was carried out as previously described [33]. The plasmids include: the constructs for the AAV7 serotype, the AAV transfer plasmid encoding either the human A53T mutant αSyn, the enhanced green fluorescent protein (GFP), human WT Rab7 with a double N-terminal HA-tag or T22N DN Rab7 with a double N-terminal HA-tag, under the control of the CMVie enhanced synapsin1 promoter and the pAdvDeltaF6 adenoviral helper plasmid. Real-time PCR analysis was used for genomic copy (GC) determination.
Primary neuronal culture
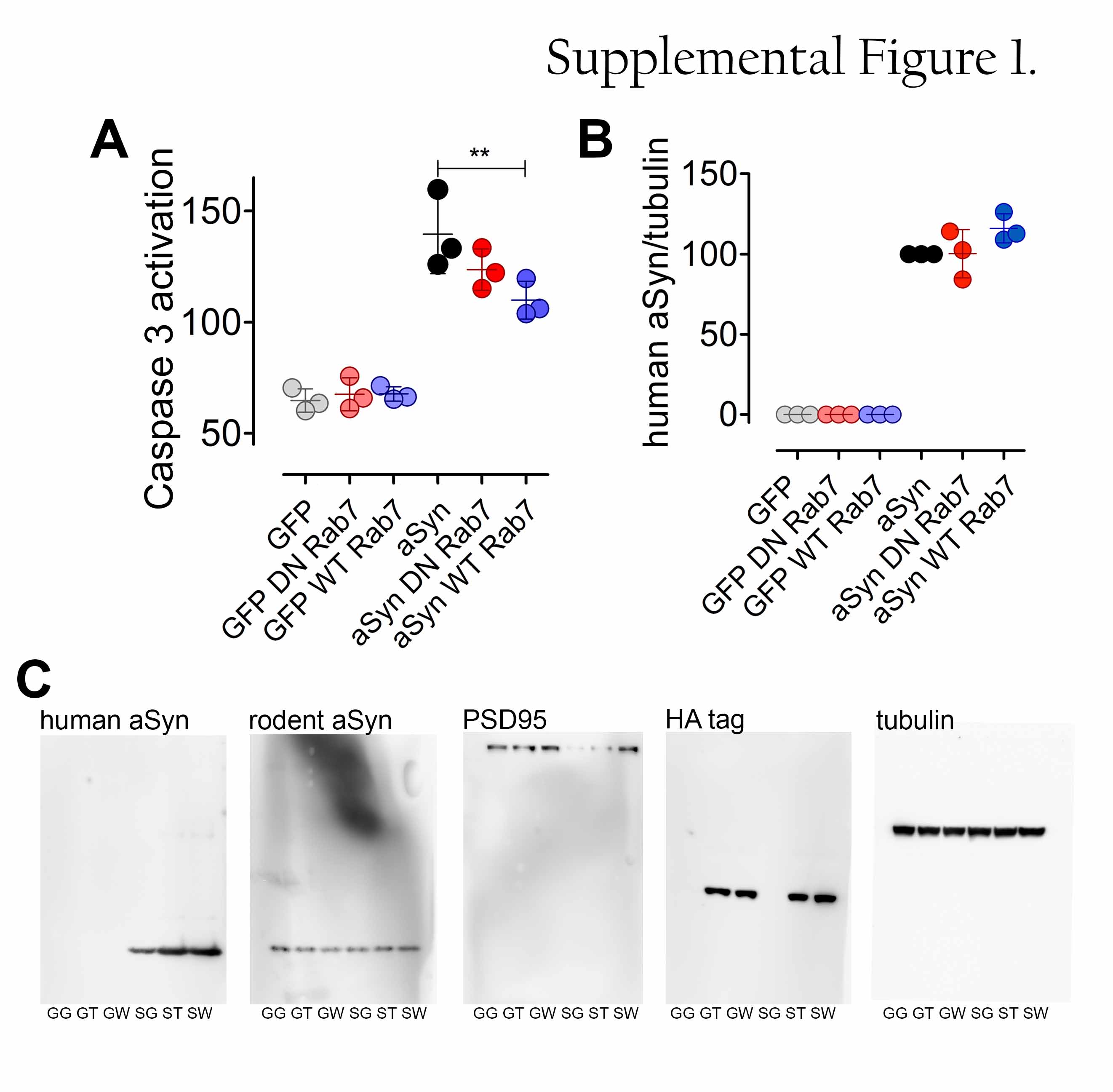
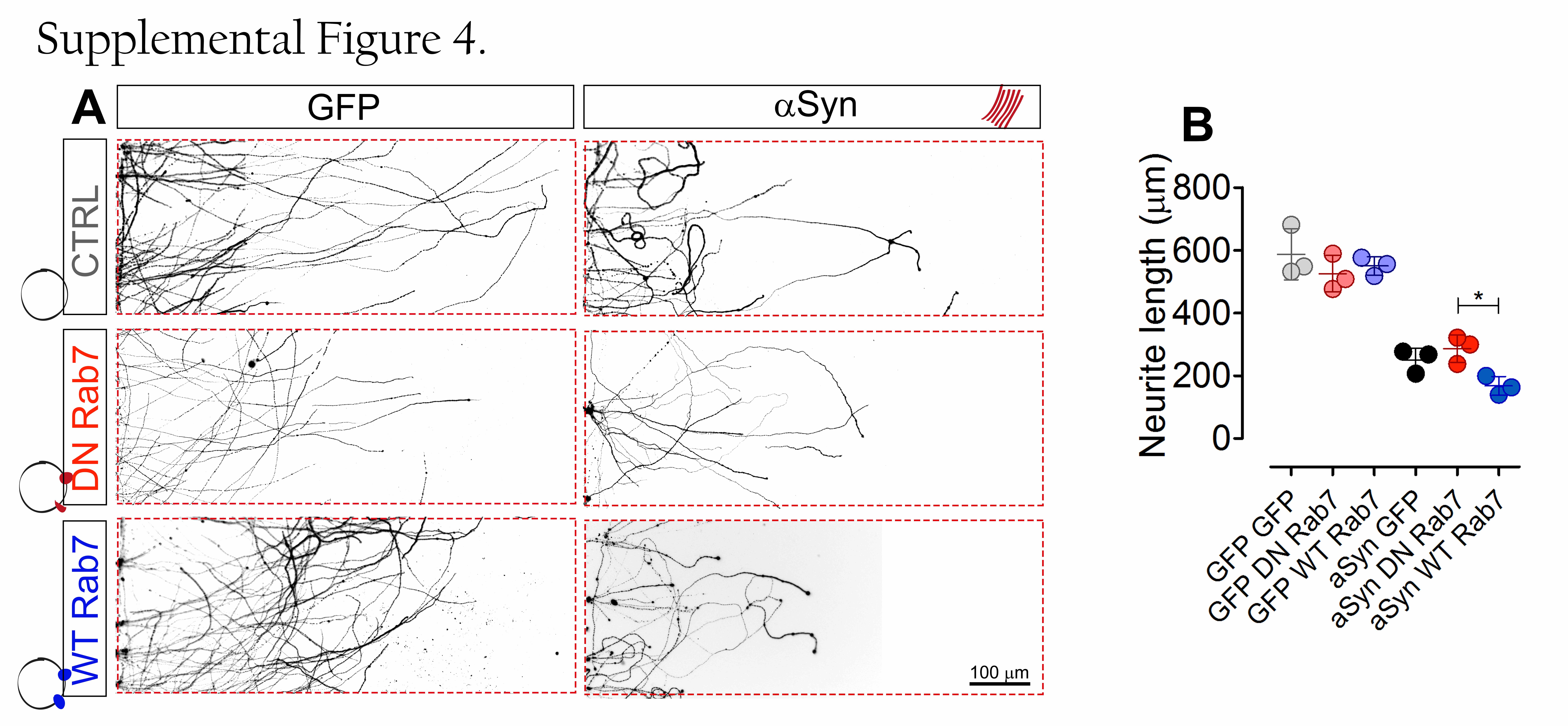
Primary cortical cultures were prepared from P1-3 C57BL6/J mouse pups of mixed sex as previously described [1]. Briefly, after dissection and trypsination, dissociated neurons were plated onto poly-L-ornithine coated glass coverslip (100 000 cells per well in 24-well dishes), and maintained in Neurobasal A medium containing 2 % B27, 0.5 mM glutamax and antibiotics. One third of the medium was changed on every third day, and from the second medium change on, no antibiotics were added. On DIV 12, neurons were transduced with one of the following combinations of rAAV2/7 vectors: (1) GFP, (2) GFP with DN Rab7A, (3) GFP with WT Rab7A, (4) A53T αSyn with GFP, (5) A53T αSyn with DN Rab7A or (6) A53T αSyn with WT Rab7A (final concentration for all 1.0e+9 GC/ml). Cells were fixed (4 % paraformaldehyde, 10 min, 20 °C) on DIV 15 for phospho-αSyn and LAMP1 labeling. Cells were fixed on DIV 16 for detecting 53BP1. Cells were fixed or lysed on DIV 18 or DIV 20 for toxicity and comet assays.
Microfluidic chambers for axonal regeneration assay
The effect of WT and DN Rab7 on αSyn-induced inhibition of axonal regeneration was measured as previously described [35]. Primary cortical neurons were seeded in silicone microfluidic devices with 450 µm groove length according to the manufacturer`s protocol. After bonding the microfluidic devices to a glass coverslip pre-coated with poly-L-ornithine, 100 000 cells (5.0e+6/ml) were plated in the cell compartment. The axonal side of the chamber was filled with 200 µl complete Neurobasal medium. After attachment of the cells, 200 µl medium was added in the cell compartment as well. One third of the medium was changed every third day on both sides. On DIV 12, neurons were infected with rAAV (final concentration for all 5e+8 GC/ml). On DIV 15, axons were mechanically cut by an air bubble. Cells were fixed on DIV 20 and stained for tubulin (PRB-435P, followed by Alexa 555-labeled anti-mouse secondary antibody). Axonal length in the distal compartment was measured using ImageJ by drawing a segmented line along each individual axon projecting out of the microgroove.
Animals and surgery
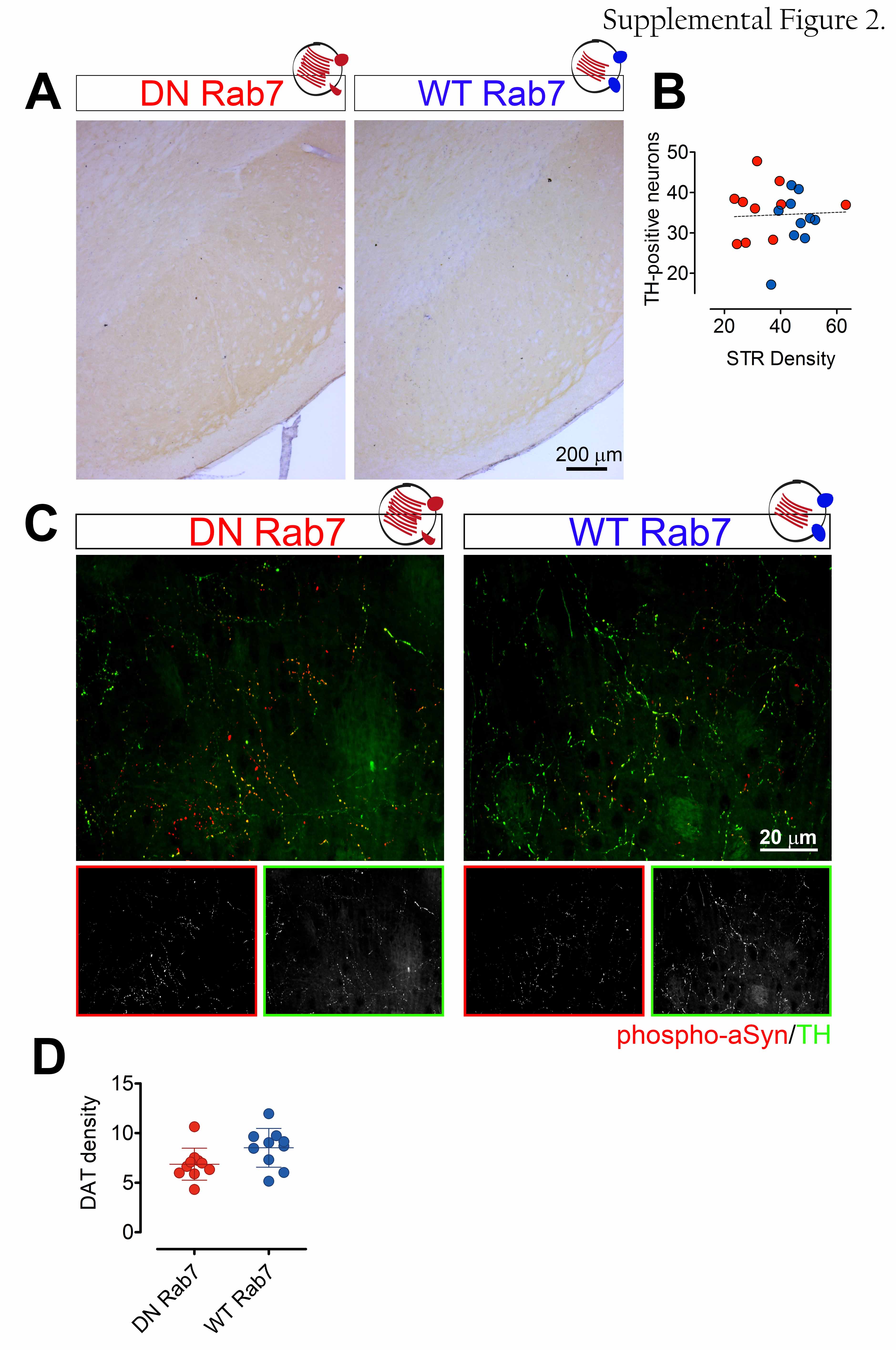
All animal experiments were carried out in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC) and approved by the Bioethical Committee of the KU Leuven (ECD project P067-2013 and P085-2014, Belgium). Eight-week-old female Wistar rats (Janvier, France, 200-250 g) were housed under a 12-hour light and/or dark cycle with free access to pelleted food and tap water. All surgical procedures were performed using aseptic techniques. Rats were anaesthetized with ketamine (60 mg/kg, intraperitoneal (i.p.), Ketalar, Pfizer, Belgium) and medetomidine (0.4 mg/kg, i.p., Dormitor, Pfizer). 3 µl of vector mix containing the rAAV2/7 A53T αSyn vector (9.0E11 GC/ml, injected genome copies 1,35E9) and rAAV2/7 HA-Rab7A T22N vector (9,3E11 GC/ml, injected genome copies 1,39E9) or the rAAV2/7 HA-Rab7A WT vector (9,3E11 GC/ml, injected genome copies 1,39E9) was injected in the substantia nigra (AP: -5.3; L: -2.0; DV: -7.2 from dura, using Bregma as reference) with a Hamilton syringe (Hamilton, Bonaduz, GR, Switzerland) as previously described [32,36].
Forelimb use was quantified by the cylinder test four weeks after the unilateral rAAV-injection. Contacts made by each forepaw with the wall of a 20-cm-wide clear glass cylinder were scored from the videotapes by an observer blinded to the animal’s identity. A total of 25 contacts were recorded for each animal, and the impaired (left) forelimb usage was expressed as a percentage of total forelimb contacts. Non-lesioned control rats score around 50 % in this test.
Rats were sacrificed four weeks after the rAAV-injection with an overdose of sodium pentobarbital (200 mg/kg, i.p.) followed by intracardial perfusion with 4 % paraformaldehyde in PBS. After post-fixation (4 % paraformaldehyde, overnight), 50 µm-thick coronal brain sections were cut with a vibrating microtome.
Immunohistochemical and immunofluorescent stainings
To quantify the number of dopaminergic neurons in the substantia nigra (SN) and the density of dopaminergic axon terminals (fibers) in the striatum, free-floating sections were stained for tyrosine hydroxylase (TH, Ab152). αSyn pathology was quantified after staining for αSyn phosphorylated at Serine 129 (phospho-αSyn, 11A5). Sections were pretreated with 3 % hydrogen peroxide (10 min) and incubated with primary antibody in 10 % normal rabbit serum overnight. Incubation with biotinylated anti-rabbit IgG (TH) and anti-mouse IgG (phospho-αSyn) secondary antibodies was followed by streptavidin - horseradish peroxidase complex. TH immunoreactivity was visualized using Vector SG as a chromogen and phospho-αSyn immunoreactivity was visualized using 3,3-diaminobenzidine (DAB).
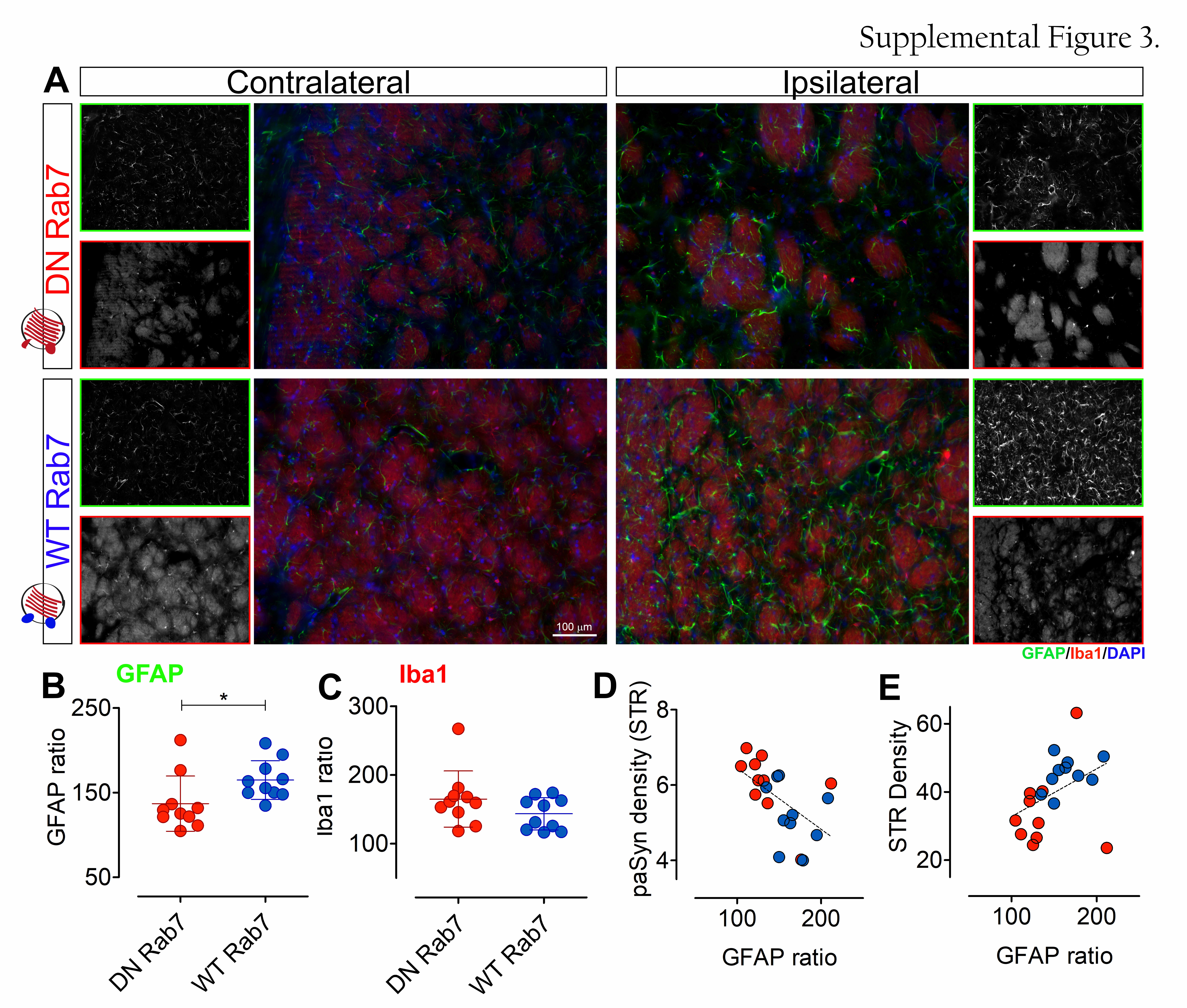
To determine neuroinflammation, every sixth striatal section was stained for the astroglia marker Glial fibrillary acidic protein (GFAP, ab4674) and for the microglia marker Iba1 (019-19741) by incubation with primary antibodies at 4 °C overnight and fluorescently labelled secondary antibodies (Alexa 488 conjugated goat anti-chicken and Alexa 555 conjugated donkey anti-rabbit, 120 min at 20 °C). Sections were counterstained with Hoechst and mounted with Fluoromount-G.
To detect DNA damage, brain slices from the SN (minimum 3 sections per animal) were stained for 53BP1 (PA5-54565), human αSyn (ALX-804-258-L001) and the HA-tag of Rab7 (MMS-101P). After incubation with secondary antibodies (Alexa 488 conjugated anti-rat, Alexa 555 conjugated anti-rabbit, and Alexa 405 conjugated anti-mouse), sections were also mounted with Fluoromount-G.
Fixed primary neurons were permeabilized using 0.2 % Triton X-100 in Tris-buffered saline (TBS, pH 7.4, 10 min). Non-specific sites were blocked by 2 % bovine serum albumin in TBS (RT, 1 hr). The following primary antibodies were applied in blocking solution (4 °C, overnight): Microtubule-associated protein 2 (MAP2, ab5392), Lysosomal-associated membrane protein 1 (LAMP1, ab208943), 53BP1, human αSyn and HA-tag (as above). Secondary antibodies were: Alexa 647 conjugated anti-chicken, Alexa 555 conjugated anti-rabbit, Alexa 488 conjugated anti-rat and Alexa 405 conjugate anti-mouse. Coverslips were mounted with Fluoromount G. Co-occurance of phospho-αSyn and LAMP1 signal was determined by using the Manders` coefficient (ImageJ), as fraction of phospho-αSyn signal within LAMP1 signal as previously [1,37].
Analysis of dopaminergic neurons, αSyn pathology and gliosis
The number of cells in the SN positive for TH and phospho-αSyn was determined by stereological measurements using the Optical fractionator method in a computerized system as described before [38] (StereoInvestigator). Every fifth section throughout the entire SN was analyzed, 7 sections for each animal. The coefficient of error calculated according to the procedure of Schmitz and Hof [39], varied between 0.05 and 0.10. The volume of striatal lesion was determined by quantifying the TH-positive terminals using the Cavalieri method (Stereologer). Every fifth section covering the entire extent of the striatum was included in the counting procedure, 7 sections for each animal. An investigator blinded to the different groups performed all the analyses.
For quantification of gliosis, fluorescent images were acquired from slices stained for GFAP and Iba1 using a 20x objective (NA 0.8) with an Axio Imager 2 microscope (Zeiss). After thresholding individual images (separately for GFAP and Iba1 channels), the area fraction was determined by ImageJ from 3-5 sections per animal and from 10 images per section as described previously [40].
For quantification of striatal phospho-αSyn pathology, images from 3-diaminobenzidine stained sections (minimum 4 sections per animal, and minimum 4 images per section) were acquired using a 20x objective (NA 0.8) with an Axio Imager 2 microscope (Zeiss). 4 independent blinded investigators rated the pattern of phospho-αSyn signal in 40-60 randomly selected images per evaluator. Blinding was achieved by 5 digit codes. Images were rated with 0 when mostly longer segments were present, and with 1 when a punctate pattern or dotted lines dominated the image. The results were analyzed in a hierarchical nested design as previously [41]. The evaluator was set as a random factor in the generalized linear mixed model, and it had no effect on the result. From the same images, the amount of phospho-αSyn staining was quantified as area fraction of the DAB signal as described for GFAP and Iba1.
Triton X-100 solubility
For αSyn protein quantification and detection of Triton X-100 insoluble proteins, neurons were lysed in buffer containing 1 % Triton X-100, 25 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA and protease inhibitors as previously described [1]. After centrifugation (14000 g, 30 min, 4 °C), supernatant was used as Triton X-100 soluble fraction. The pellet (Triton X-100 insoluble fraction) was washed in ice-cold PBS, centrifuged again and re-dissolved with sonication (10 s) in 50 µl buffer containing 2 % SDS, 75 mM Tris, 15 % glycerin, 3.75 mM EDTA pH 7.4 and protease inhibitors. 10 µg of Triton X-100 soluble lysate or 10 µl from the re-dissolved pellet were loaded onto a 4-20 % Tris/glycine SDS gel for western blot analysis. After blocking, membranes were incubated first in the presence of antibodies against human αSyn (ALX-804-258-L001), HA tag (MMS-101P), PSD-95 (3450) and alpha-tubulin (ab6046), then with horse radish peroxidase-conjugated secondary antibodies (donkey anti-rat, donkey anti-mouse and donkey anti-rabbit). Signal was visualized with chemiluminescent substrate and detected with Luminescent Image Analyzer.
Biochemical assays
To determine the toxic effect of αSyn in cultured neurons, the concentration of LDH was measured in the medium 24 h after the last medium change using the Cytotoxicity Detection Kit (Roche) according to the manufacturer’s protocol. Briefly, for each independent preparations, 3-4 technical replicates were used and averaged. Absorbances were measured at 492 nm, reference wavelength was 620 nm. Background (medium) absorbance was substracted from all values, and values were normalized to control (non-treated) and to maximal lysed (treated with 2 % Triton X-100, according to the protocol) cells. Caspase activity was measured with EnzChek Caspase-3 Assay Kit (Thermo Fisher). Apoptosis was induced with 100 µM NMDA for 24 h. Cells were lysed and protein concentration determined (BCA assay). 100 µM Z-DEVD–AMC substrate was added to a sample of 100 µg protein. After 45 min incubation at 20 °C, fluorescence of cleaved Z-DEVD–AMC was acquired (Excitation/Emission: 342/441 nm; Infinite 200 Pro). MAP2 stained primary neurons were quantified as area fraction from 20x images as described for GFAP and Iba1.
Mitochondrial membrane potential, cytoplasmic reactive oxygen species (ROS) production and mitochondrial ROS production were measured in separate assays as described previously [1]. Tetraethylbenzimidazolylcarbocyanine iodide (JC-1, 50 nM, 37 °C, 30 min) was used to measure the mitochondrial membrane potential. Fluorescence at 485/505 nm (excitation/emission) and 535/570 nm was detected by a plate reader (Infinite 200 Pro). After measuring the basal mitochondrial membrane potential, the mitochondrial uncoupler FCCP (1 mM) was used to induce a mitochondrial stress.
Cytoplasmic ROS was measured using dichlorodihydrofluorescein diacetate (DCFH-DA; 10 mM, 37 C, 30 min), mitochondrial ROS was measured using MitoSOX (200 nM, 20 min, 37C). Fluorescence at 485/530 nm (DCFH-DA) or 510/580 nm (MitoSox) was measured with plate reader. To detect the response to stress-induced ROS production, preparations were treated with 1 mM FCCP (MitoSox) or with 1 % H2O2 (DCFH-DA).
DNA damage
To determine DNA double strand breaks, the number of 53BP1-positive nuclear loci was counted in brain sections containing nigral neurons transduced with human αSyn (minimum 3 sections per animal, and minimum 3 images per section, 15-30 neurons per animal) or in primary neuronal cultures (3 repetitions, minimum 15 neurons per repetition /condition) as previously described [18].
DNA single- and double-strand breaks were detected via single cell gel electrophoresis (comet) assay, according to the guidelines provided in [42]. Adherent microscopy slides were coated with 1 % low melting agarose (PeqLab). Primary neurons were harvested in PBS, mixed with 1 % agarose in PBS and placed on the pre-coated slide. After agarose gelling, slides were submerged in lysis buffer (2.5 M NaCl, 100 mM Na2EDTA, 0.1 % Triton X-100, 10 mM TRIS, pH 10, 4 °C, 1 h) and denatured in alkaline electrophoresis buffer (0.3 M NaOH, 1 mM Na2EDTA, pH>13, 30 min). Electrophoresis was carried out for 25 min at 20 V in the same buffer. Slides were removed from the electrophoresis chamber, fixed in 70 % ethanol and stained with 2.5 μg/ml of propidium iodide for 20 min at 20 °C. Excess of stain was removed by rinsing the slides with distilled water. For each slide, at least 10 images were acquired using an epifluorescence microscope (Zeiss Axio Observer). For each comet, the “tail moment” was determined as the product of the percentage of DNA in the tail and the tail length using the Open Comet plugin for Image J as described previously [17].
Statistical analysis
For in vitro experiments, “n” was set to the number of individual preparations. Within one experiment, mean of the technical replicates was determined for each treatment group. In animal experiments, “n” was set to the number of animals. No animals were excluded from the analysis. In graphs, markers represent individual preparations or individual animals, lines represent mean and standard deviation (SD) of all individual preparations or all animals. Data normality was tested by the Shapiro-Wilk test and graphically by QQ plot (R). t-test, one-way ANOVA or two-way ANOVA were performed using GraphPad Prism. Linear regression was performed using R (v 2.8.0). Binomial data from pattern rating was analyzed using generalized linear mixed-effects model fitted with binomial distribution. Evaluator, animal, section and image were used as random effects nested within each other (R package: lme4). P values are indicated in the graphs by symbols with * or # representing p<0.05, ** or ## representing p<0.01, *** or ### representing p<0.001. Exact p values are given in the Figure legend.
{kind=link}
{kind=link}
{kind=link}
{kind=link}