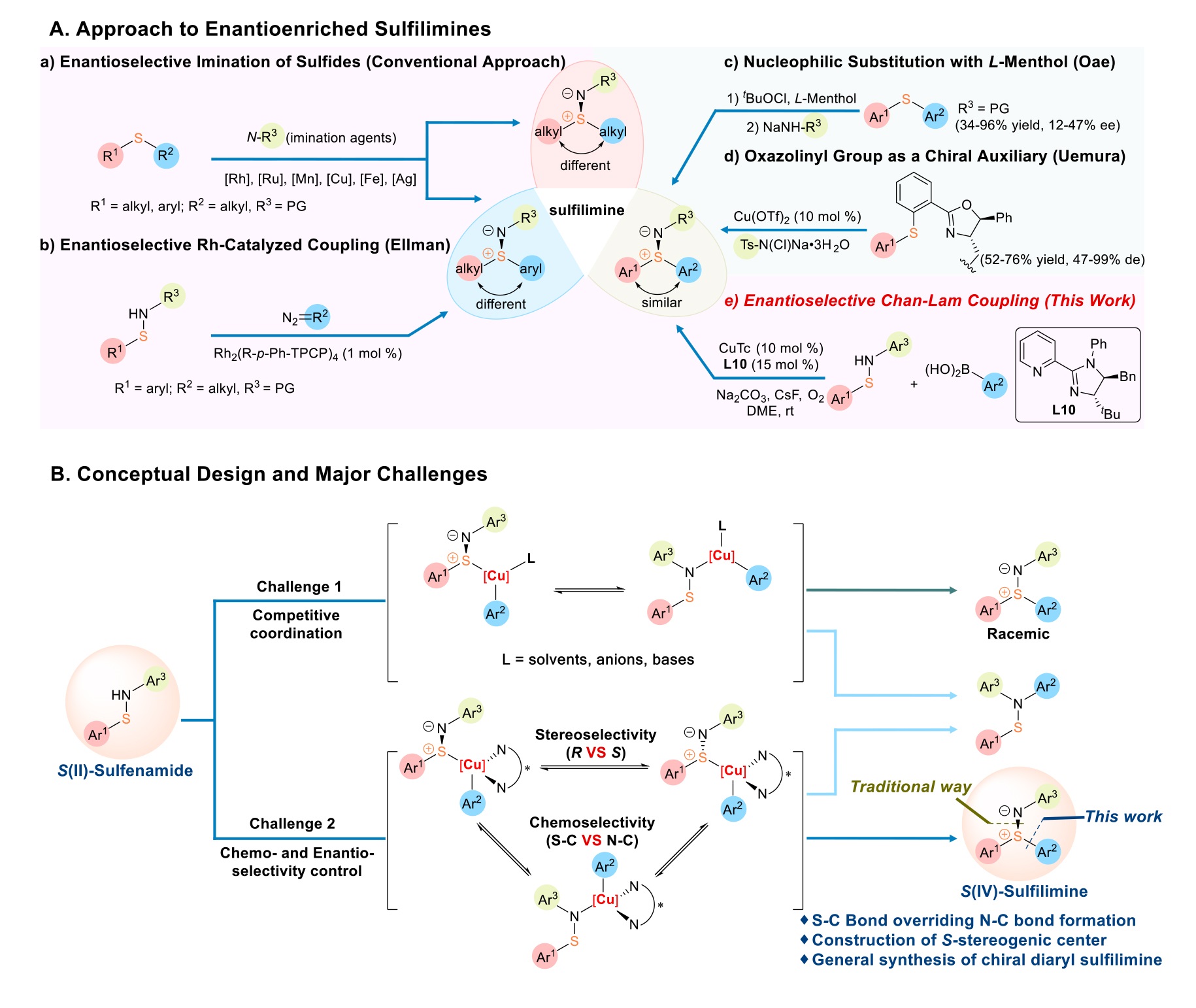
Reaction development. Control of the copper coordination sphere is the key to realizing ligand-controlled enantioselective Chan-Lam coupling. Reasoning, that a chelating substrate combined with a bidentate ligand would likely inhibit transmetallation from the aryl boronic acid, chelating N-groups such as acyl, carbamoyl, etc. were not employed. Rather, we strategically chose a sulfenamide bearing a phenyl group on nitrogen (1a) as model substrate, along with 4-tert-butyl phenylboronic acid (2a), for examination of the process. Initially, a series of privileged nitrogen-based ligands were surveyed in the presence of a copper catalyst and amine base to quench the boronic acid byproduct (Table 1, Table S1 and Table S5). Among the chiral scaffolds tested, 2-pyridyl oxazolidine ligand L4 outperformed by delivering the desired product 3aa in 43% ee (Table 1, entry 4). Further optimization of the other parameters with this ligand (see Table S2-4) revealed that iPr2NEt (2.0 equiv) as base, dimethoxyethane (DME) as solvent, and copper(I) thiophene-2-carboxylate (CuTc, 10 mol %) as catalysts improved the outcome forming 3aa with 50% yield and 58% ee (entry 5). Perturbing the ligand to a slightly nonplanar geometry by incorporation of an aryl group in L5 (entry 6) improved the assay yield (60%) and enantiomeric excess (71%) of 3aa. Permutation of this substitution to the imidazole moiety (L6) leads to superior enantiocontrol (80% ee) albeit with slightly diminished assay yield (44%, entry 7). Installation of a further ortho-fluoro group on the pyridine (L7) led to much lower yield and ee values (entry 8). Consequently, further modification of the ligands focused on the imidazole portion. At this juncture, addition of CsF (50 mol %) was examined to enhance turnover by activation of the boronic acid31. In line with this hypothesis, the yield was doubled while retaining the same enantioselectivity (entry 9 vs entry 7). As such, was CsF employed in all further trials. Addition of an ethyl group to the 5-poistion of the imidazole ring would further constrain the geometry and shift the tBu closer to bound substrates. Indeed, this perturbation resulted in excellent catalytic activity (85–86% assay yield) with the trans disposition in L9 giving the higher enantioselectivity (90% ee; compare entry 11 with 10). Further increasing the steric effect from the 5-position by employing a benzyl group in place of the ethyl group (L10) resulted in improved enantioselectivity (92% ee) while maintaining the high efficiency of the transformation. Ultimately, adjusting the base from iPr2NEt to Na2CO3 afforded 3aa in 88% assay yield, 83% isolated yield, with 92% ee (entry 11). Thus, the optimal conditions for enantioselective Chan-Lam coupling of sulfenamide were determined to be: 1a as limiting reagent, 2a (2.0 equiv) as coupling partner, CuTc (10 mol %)/L10 (15 mol %) as catalyst system, Na2CO3 (2.0 equiv) as base, CsF (50 mol %) as additive, in DME under an O2 atmosphere at room temperature for 12 h. For a complete list of optimization conditions, see Tables S1-9 in SI for details.
Substrate Scope. With the optimal conditions established, we examined the generality of this enantioselective Chan-Lam coupling reaction (Table 2). The larger naphthalene substrate (1b) was similarly effective and a crystal structure thereof permitted unambiguous assignment of the absolute (S)-configuration (see Table S10-16 for details); the configurations of the other products in Table 2 were assigned by analogy. Electron-donating 4-OMe (1c) and 4-Me (1d) or electron-withdrawing 4-F (1a), 4-Cl (1e), 4-Br (1f), 4-CO2Et (1g), 4-CF3 (1h), 4-CN (1i), and 4-NO2 (1j) aryl groups on the sulfur of the sulfenamide were all well tolerated furnishing the corresponding products in good yields with excellent enantiocontrol. Attesting to the mild conditions, various functional groups, such as aldehyde (1k), ketone (1l), ester (1g), amide (1m) and alcohol (1n) were all compatible under the optimal conditions, providing the products in 61 − 72% yields and 81 − 91% ee. Remarkably, this enantioselective Chan-Lam coupling protocol displayed excellent chemoselectivity favoring S-arylation of sulfenamide (1a) over other functional groups that typically undergo Chan-Lam coupling such as the amide N-H (1m) or alcohol O-H (1n), highlighting the unique nature of this protocol. The chemistry was well-accommodated with sulfenamides bearing meta-substituted aryl groups, as evidenced by the formation of 3oa and 3pa in 67% with 89% ee and 73% with 88% ee, respectively. Moreover, the newly devised method proceeded smoothly with challenging S-heterocyclic sulfenamides allowing an array of chiral S-heteroaryl sulfilimines, including pyridyl (3qa, 3ra), quinolinyl (3sa), and thienyl (3ta, 3ua) to be obtained with good enantioselectivities (70–88% ee), albeit in modest yields (45 − 75%).
Next, the N-aryl substitution of the sulfenamides was examined. Both electron-donating (1v) and electron-withdrawing aryls (1w, 1x, 1y) gave the desired products (3va-ya) with 60–84% yields and 83–94% enantioselectivity. Importantly, a set of functional groups, including aldehyde (1z), ketone (1aa), ester (1y), and even the polymerizable vinyl group (1ab), were well tolerated in the protocol to deliver the corresponding chiral diaryl sulfilimines (3ya-aba) in good yields (77 − 89%) with good to excellent enantiocontrol (74 − 93% ee). These results highlight the advantages of this protocol as the aldehyde and terminal alkene are not tolerated in the previously reported route, oxidative imination of sulfides, due to the competing oxidation. Sterically hindered N-(2-Br-phenyl) sulfenamide (1ac) reacted with 2a under the optimal conditions to afford 3aca in 74% with 84% ee. meta-Substituted N-aryl sulfenamides, including 3-methoxy (1ad), 3-iodo (1ae), and 3-acetamido (1af), were competent coupling partners to deliver sulfilimines 3ada-fa in 71 − 81% yields with 74 − 91% ee. Heteroaromatic sulfenamides, such as pyridines 1ag, 1ah and pyrimidine 1ai, also proved to be potent coupling partners to furnish chiral N-heterocyclic diaryl sulfilimines 3aga-ia in satisfactory yields with good stereocontrol.
The use of alternate arylboronic acids was next interrogated. The 2-naphthalene (2b) variant again proved effective at this position. Electron-donating substrates including 4-OBn (2c), 4-nbutyl (2d), 4-TMS (2e), 4-PhO (2f), and 4-Ph (2g) and electron-neutral substrates 4-MeS (2h) and 4-H (2i) furnished the products 3ab-i in moderate to good yields while sustaining good stereoselectivities. In particular, 3ah is synthetically intractable via the classic imination of sulfides, since it contains two functionalities with different oxidation states of sulfur. Importantly, labile trimethylsilyl (2e) or vinyl (1j) groups were also amendable to the chemistry, providing a means for later modification via other orthogonal processes. Moreover, meta-substituted arylboronic acids (2k-m) were effective. Finally, the heterocyclic benzofuranyl boronic acid (2n) generated 3an with good selectivity (87% ee).
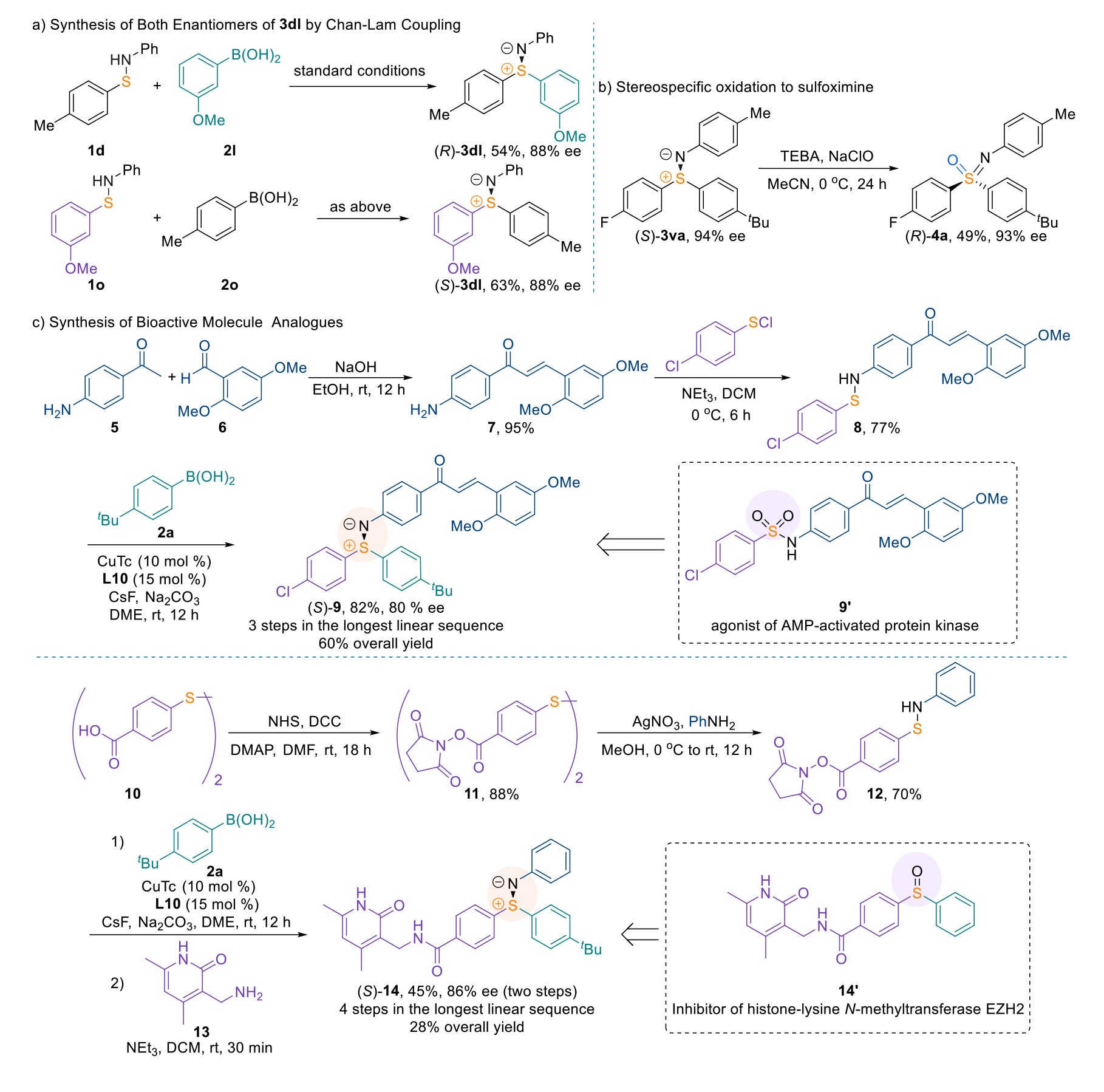
Synthetic Utility. A major source of nitrogen-based chiral ligands is inexpensive and abundant natural L-amino acids, which, in turn, limits the availability of the enantiomeric congeners. In this instance, the opposite enantiomeric ligand of L10 is derived from an expensive D-amino acid. Thus, generation of both of enantiomeric products from the same, less expensive chiral ligand would be a distinct advantage. Simply exchanging the aryl groups of the sulfenamide and arylboronic acid substrates enabled such an outcome. Thus, synthesis of both enantiomers of sulfilimine 3dl was achieved by using 1d with 2l vs 1o with 2o under the identical catalytic conditions (Scheme 2a). In addition, the chiral S(IV)-based sulfilimines obtained by our enantioselective Chan-Lam S-alkylation could serve as versatile intermediates to access to chiral S(VI)-based sulfoximines. Exposure of sulfilimine 3va to NaClO leads to the formation of sulfoximine 4a with no detectable erosion of enantiomeric excess at the sulfur stereocenter32 (Scheme 2b). Next, we showed the synthetic utility of this method due to the emerging role of sulfilimines in medicinal chemistry. Inspired by the elaboration of pan-CDK inhibitor Bay1000394 from Bayer by switching the sulfonamide to a sulfoximine as the pharmocophore33, two sulfilimine analogues of patented bioactive molecules were generated using our enantioselective Chan-Lam coupling as the key step (Scheme 2c). Sulfilimine 9, an analog of an agonist (9’) of AMP-activated protein kinase (AMPK) in the treatment of degenerative neurological diseases34, could be synthesized in three steps with 60% overall yield and 80% ee. Compound 14, an analog of the sulfoxide-based histone-lysine N-methyltransferase EZH2 Inhibitor (14’)35, was concisely assembled in four steps with a 28% yield and 86% ee. Remarkably, compound 12 bearing a labile activated ester group reacted efficiently with 2a to deliver the corresponding chiral sulfilimine in good stereoselectivity, underlining the broad functional group compatibility. Overall, the method described herein offers a versatile platform to afford a range of derivatives in a step-economic manner.
Mechanistic Studies. To understand the factors controlling chemo- and enantioselectivity, the mechanism was probed using density functional theory (DFT) [UM06/6-311 + + G(d,p)-SDD(Cu)-CPCM(DME)//UB3LYP-D3/6-31G(d)-SDD(Cu)36–44, see Supporting Information for full computational details]. Initially, CuII complex 1’ forms the pre-reacting complex 1 with the incoming aryl boronic acid (Fig. 1a, downhill in energy by 2.0 kcal/mol). This complex then undergoes transmetalation (via [1–2], 3.9 kcal/mol) to form intermediate 2 (−25.1 kcal/mol) following dissociation of boric acid. Next, sulfenamide coordinates to 2 to form 2a’ (−20.6 kcal/mol).
Intermediate 2a’ can undergo kinetically controlled S-arylation (Fig. 1b, blue pathway) or the higher energy N-arylation (Fig. 1b, red pathway). For the S-arylation, CuII intermediate 2a’ first undergoes disproportionation with CuII intermediate 1’ to form CuIII complex 2’ (Fig. 1b, blue, −22.4 kcal/mol) and LCuI hydroxide. Thiophene carboxylate dissociates from the inner coordination sphere of the CuIII complex to form the square pyramidal CuIII cationic complex 2c’. Cystallographic evidence of various square pyramidal cationic CuIII species supports the formation of such an intermediate45. Furthermore, the imidazole ring of L2, a strong sigma donor, is trans to sulfur, (see Figure S1). Next, sulfur-arylation occurs via [2–3] (overall energetic span46 of 8.0 kcal/mol from intermediate 2a’) to give protonated S-arylation product 3’-S. Finally, deprotonation of the nitrogen atom gives the final product 3-S.
To understand the chemoselectivity observed in this transformation, we also computed the formation of the N-aryl product (Fig. 1b, red). From intermediate 2a’, deprotonation must occur prior to N-arylation in order for the nitrogen atom to react (for a comparison of energetic spans of N-arylation before and after deprotonation, see Figure S2). The nitrogen atom of the sulfenamide undergoes deprotonation with NMe3 as the base (as a simplified model for iPr2NEt) via 2’-TS (overall energetic span of 14.0 kcal/mol) to form intermediate 2d’. Disproportionation of CuII species 2d’ with CuII intermediate 1’ yields CuIII intermediate 2b’ and LCuI hydroxide. From intermediate 2b’, N-arylation occurs via [2’-4], giving the thermodynamically favored product 4. Notably, S-arylation is under kinetic control as the energetic span to form the product 3’-S is lower by 6.0 kcal/mol, explaining the observed experimental chemoselectivity in this transformation. This kinetic control overcomes the far greater stability of the N-aryl product relative to the S-aryl product30.
Next, we delved further into the chemoselectivity by comparing the experimental results with different ligands to our computational results (see Figures S3-4 in the Supporting Information). Notably, ligand L47, which gives a greater amount of N-arylation product, has a smaller difference between energy spans for S-arylation and N-arylation (see Table S17). This agreement of the experimental results with our computational findings provides additional support for the proposed mechanism.
Finally, the origin of enantioselectivity in this transformation was investigated. The lowest energy conformations of the S-arylation transition state leading to (S)-enantiomer and (R)-enantiomer are shown in Fig. 1c (for comparison of energetics with different methods, see Tables S18-20). Upon initial inspection of the transition state geometries, it is unclear what factors control the excellent enantioselectivity observed in this transformation (see Figure S5). As a result, interaction/distortion analysis was performed as described by Houk and Bickelhaupt47 (see Figure S6) in which the favorable interaction energy between the copper-ligand (CuL) and aryl substrate is compared with the energy required to distort the intermediates into the transition state geometries. The distortion in the LCu component of [2–3]R is 27.2 kcal/mol, which is much greater than the analogous distortion in the [2–3]S (12.0 kcal/mol). This distortion in the LCu component can be observed in the overlaid transition state and intermediate geometries given in Figure S7. However, the interaction energy between the two LCu and aryl substrate components is greater in [2–3]R, giving an overall energy of −34.4 kcal/mol compared to −34.1 kcal/mol in [2–3]S. Based on this analysis, the observed enantioselectivity does not arise predominantly from a difference in interaction energy. Rather, the difference in distortion need to accommodate the two transition states drives the enantioselectivity.
Thus, the difference in free energy between [2–3]R and [2–3]S was further explored. Since [2–3]S is favored by 2.9 kcal/mol in free energy, but only by 0.3 kcal/mol in enthalpy, we attributed the enantioselectivity to this free energy difference. On further examination of the transition state geometries (Fig. 1c), the S-arylation leading to the (S)-enantiomer has an 8-membered ring involved in the bond formation whereas [2–3]R has a 6-membered ring. Since the 8-membered ring is more flexible, it is more entropically favored, which results in a favorable free energy for [2–3]S.
Finally, the decrease in the observed experimental enantioselectivity when the substrate is changed from the unsubstituted phenyl substrate (left, Figure S7) to the p-NO2 substituted substrate (right, Figure S7) was computed with L10 ligand. In agreement with the observed experimental decrease in enantioselectivity (from 92% ee to 56% ee), the free energy difference also decreases (from 2.9 kcal/mol to 0.7 kcal/mol).
{kind=link}
{kind=link}