Chemicals and reagents
Primary antibodies against total AKT, slug, β-actin and horseradish peroxidase-conjugated secondary antibodies were purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). Primary antibody against p-AKT Ser473 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Primary antibody against CDK6 were purchased from Proteintech Group, Inc (Wuhan, China). MK2206 and PD-0332991 were purchased from Selleck Chemicals (Houston, TX, USA).
Cell culture
The human hepatocyte-derived cell line HL7702 and HCC Huh7, BEL7402 and SK-HEP-1 cell lines were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cells were propagated in Dulbecco′s Modified Eagle Medium/F12 (DMEM/F12) containing 10% fetal bovine serum (FBS). Cells were maintained at 37 °C in a humidified incubator with 5% CO2 in air.
Western blot assay
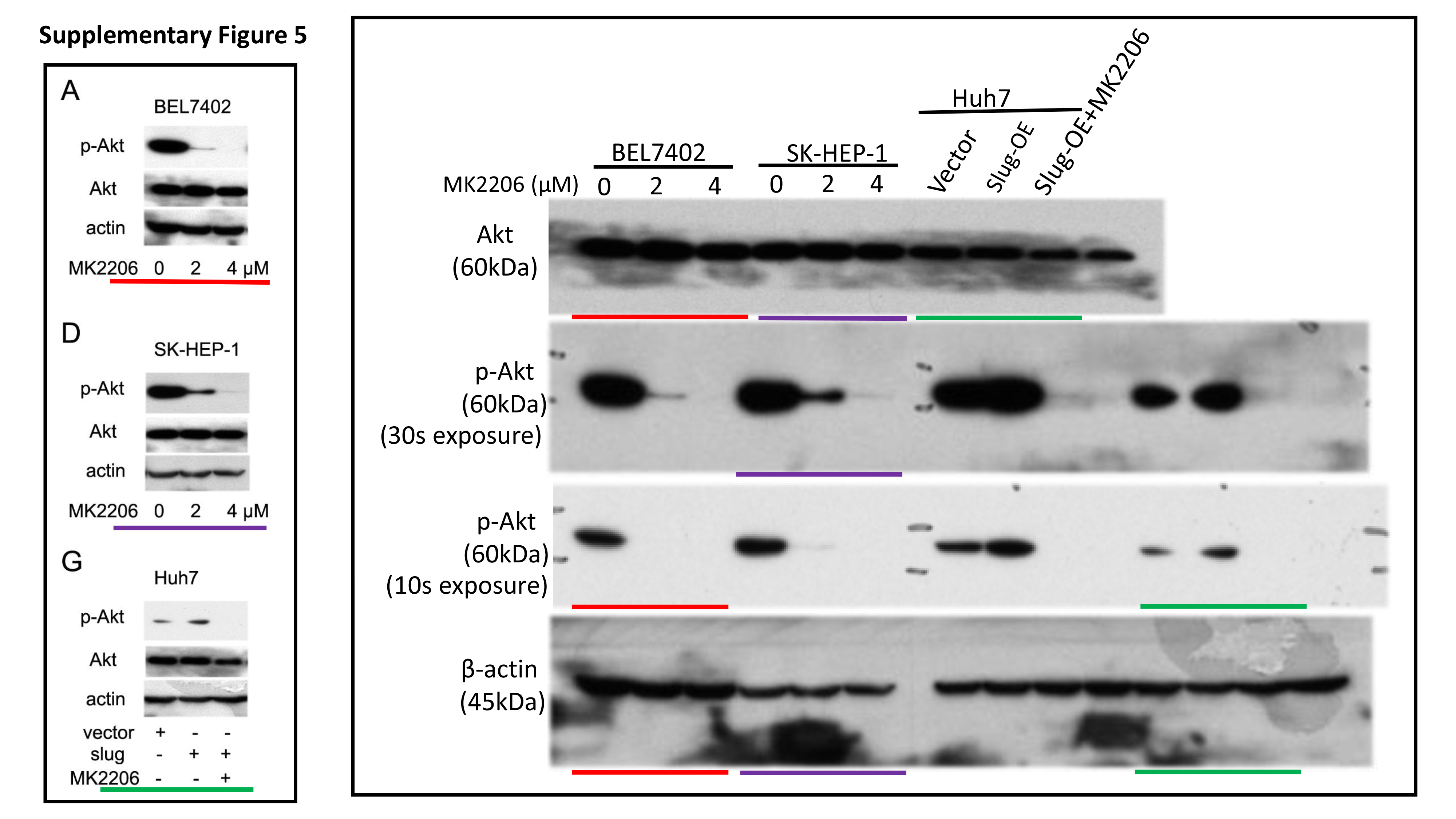
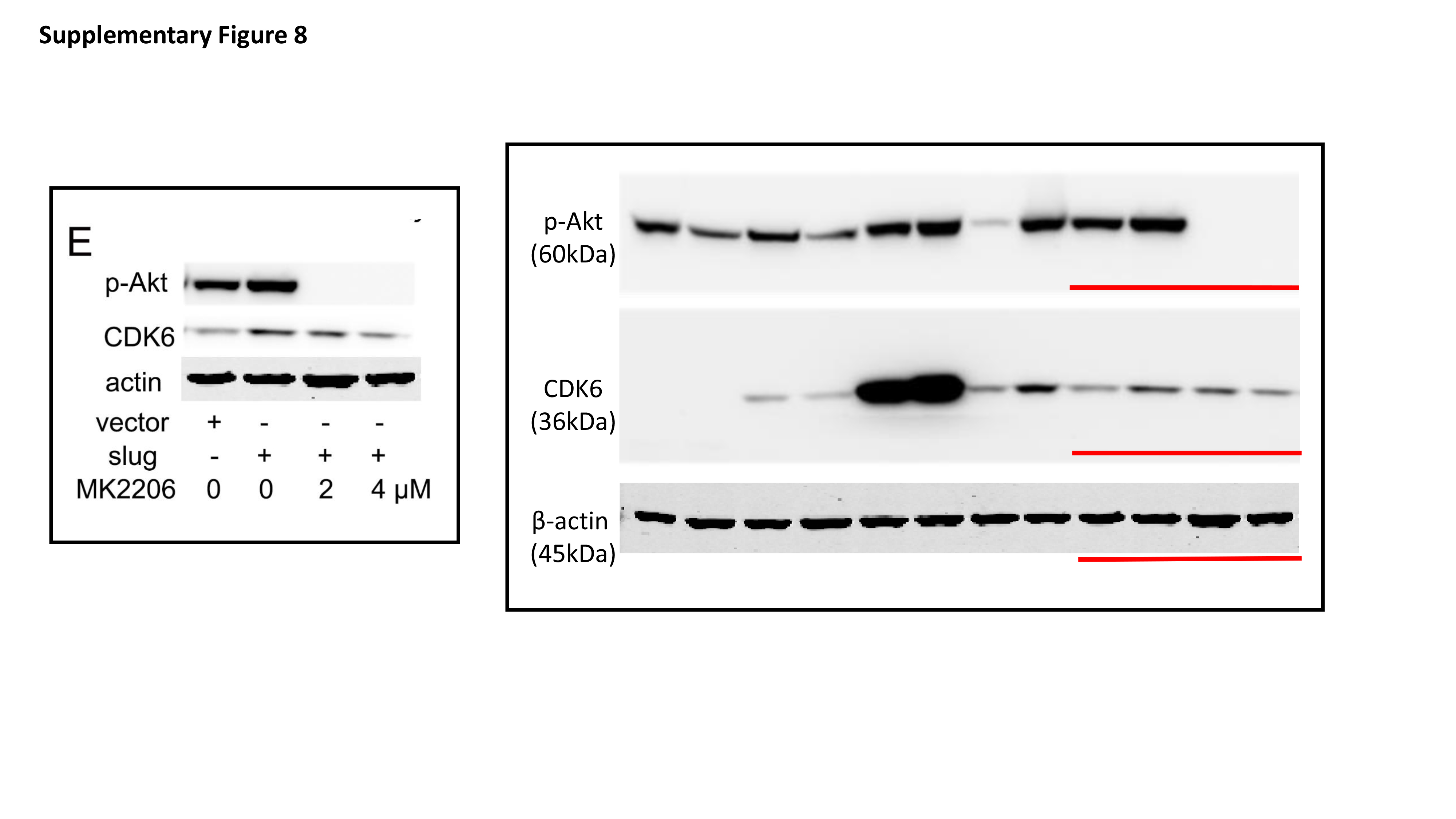
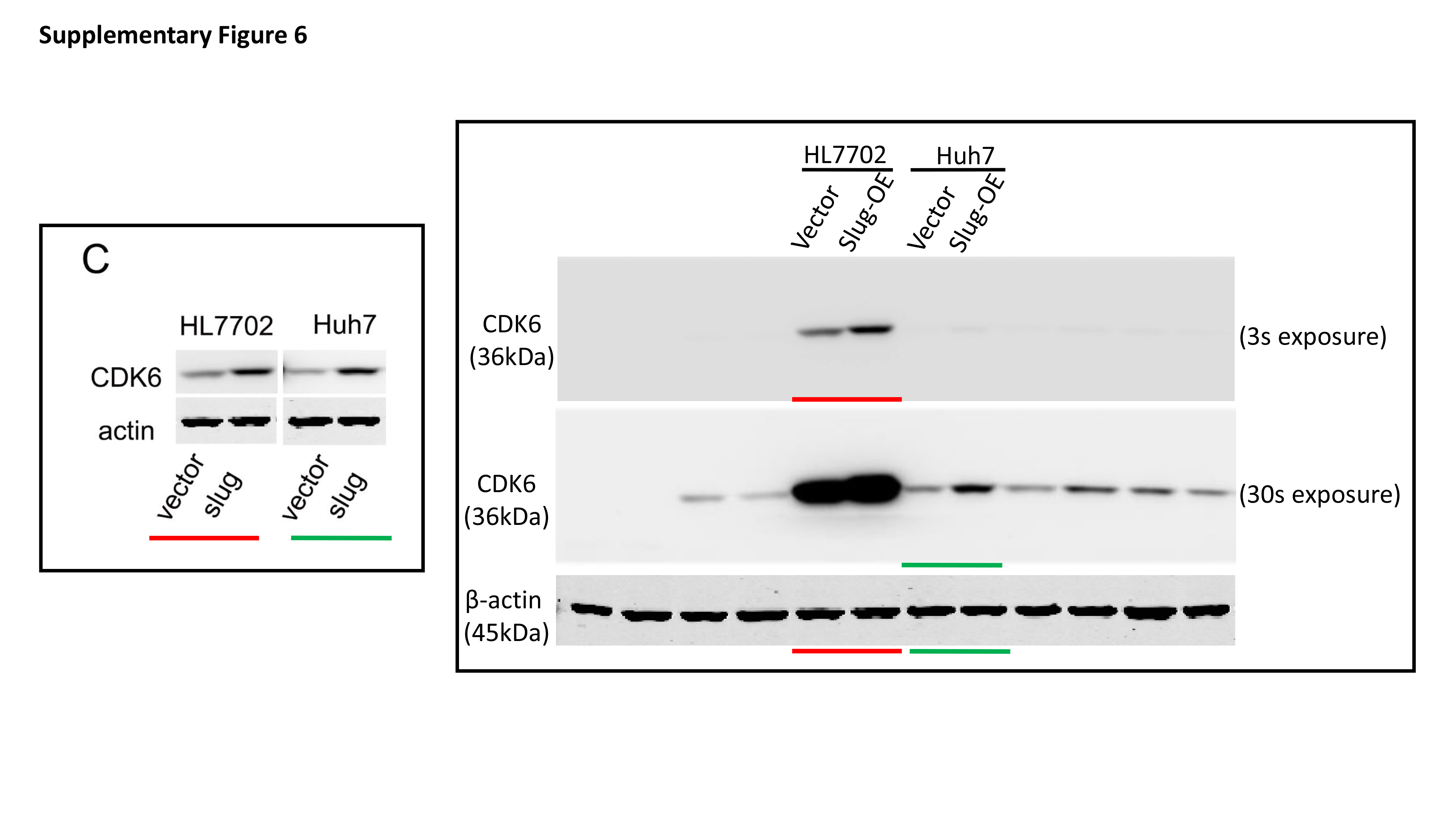
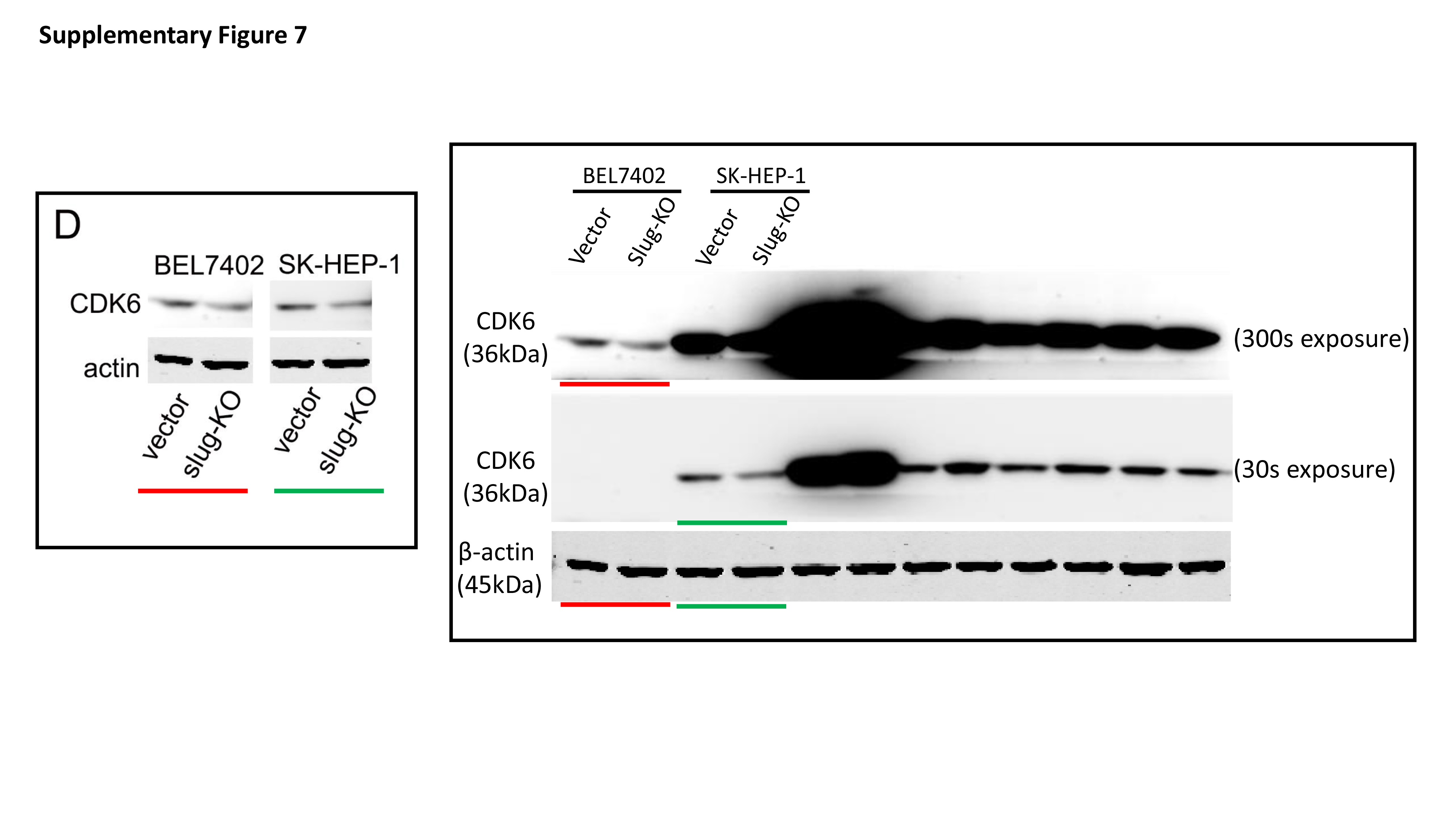
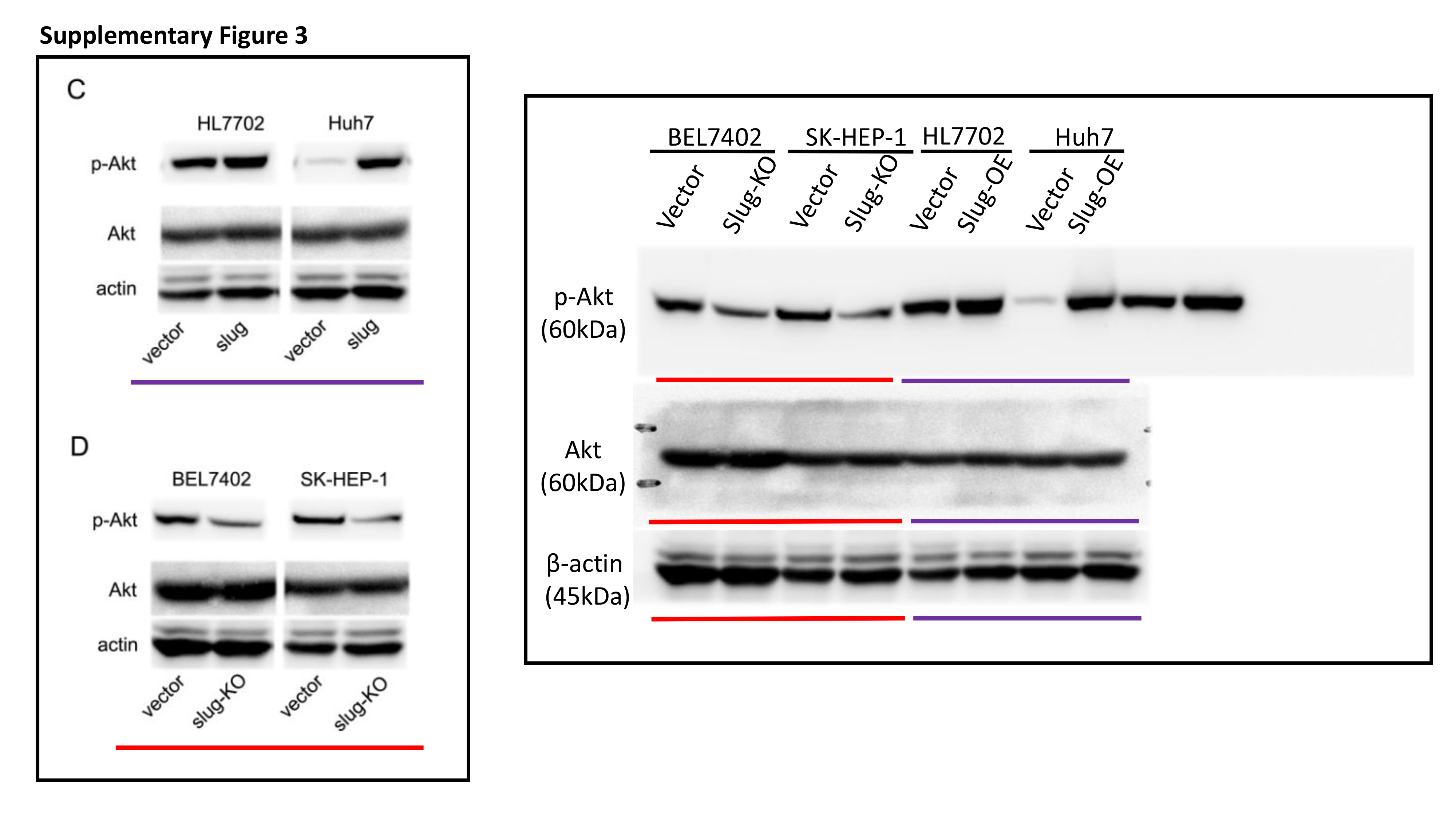
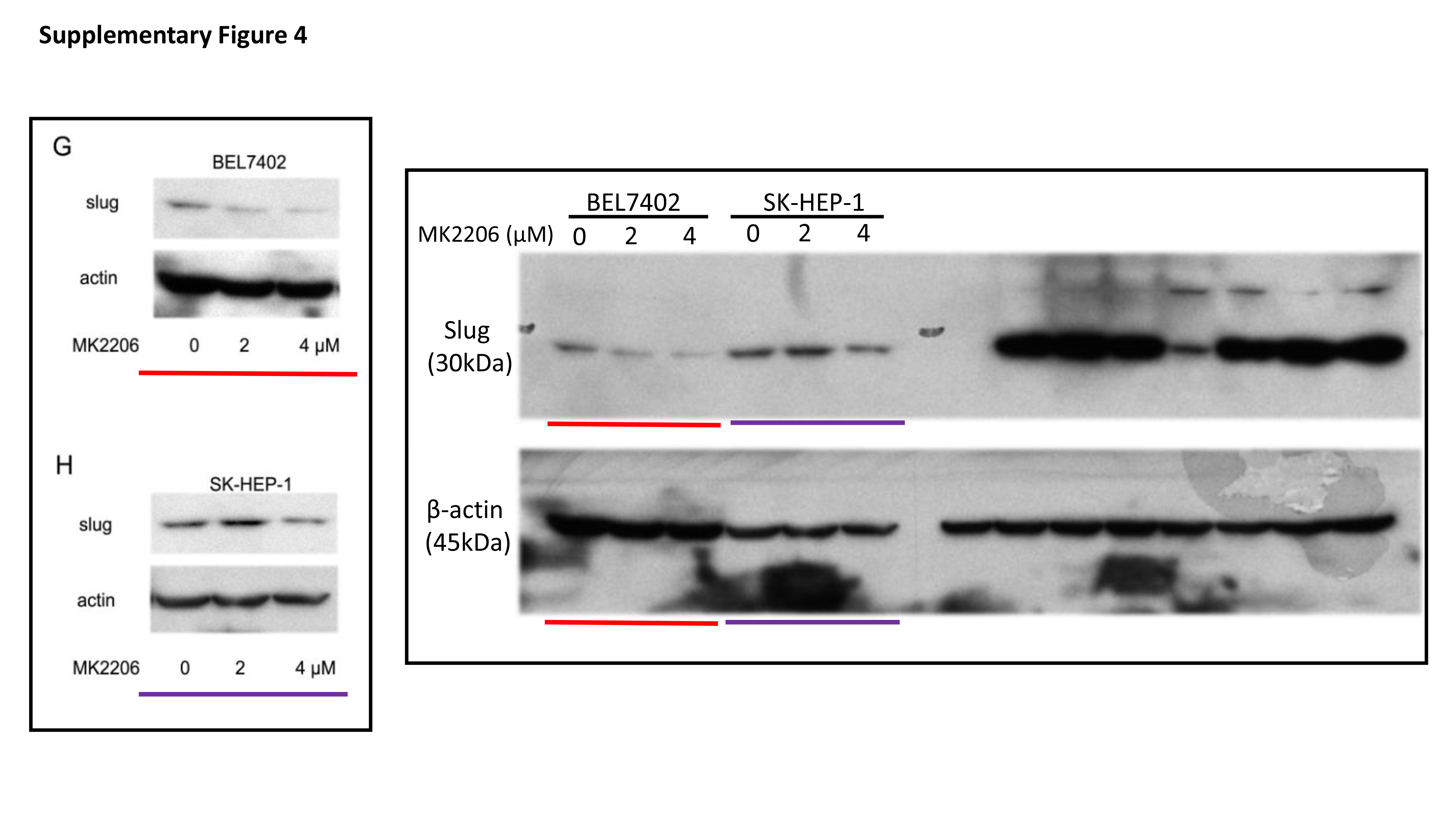
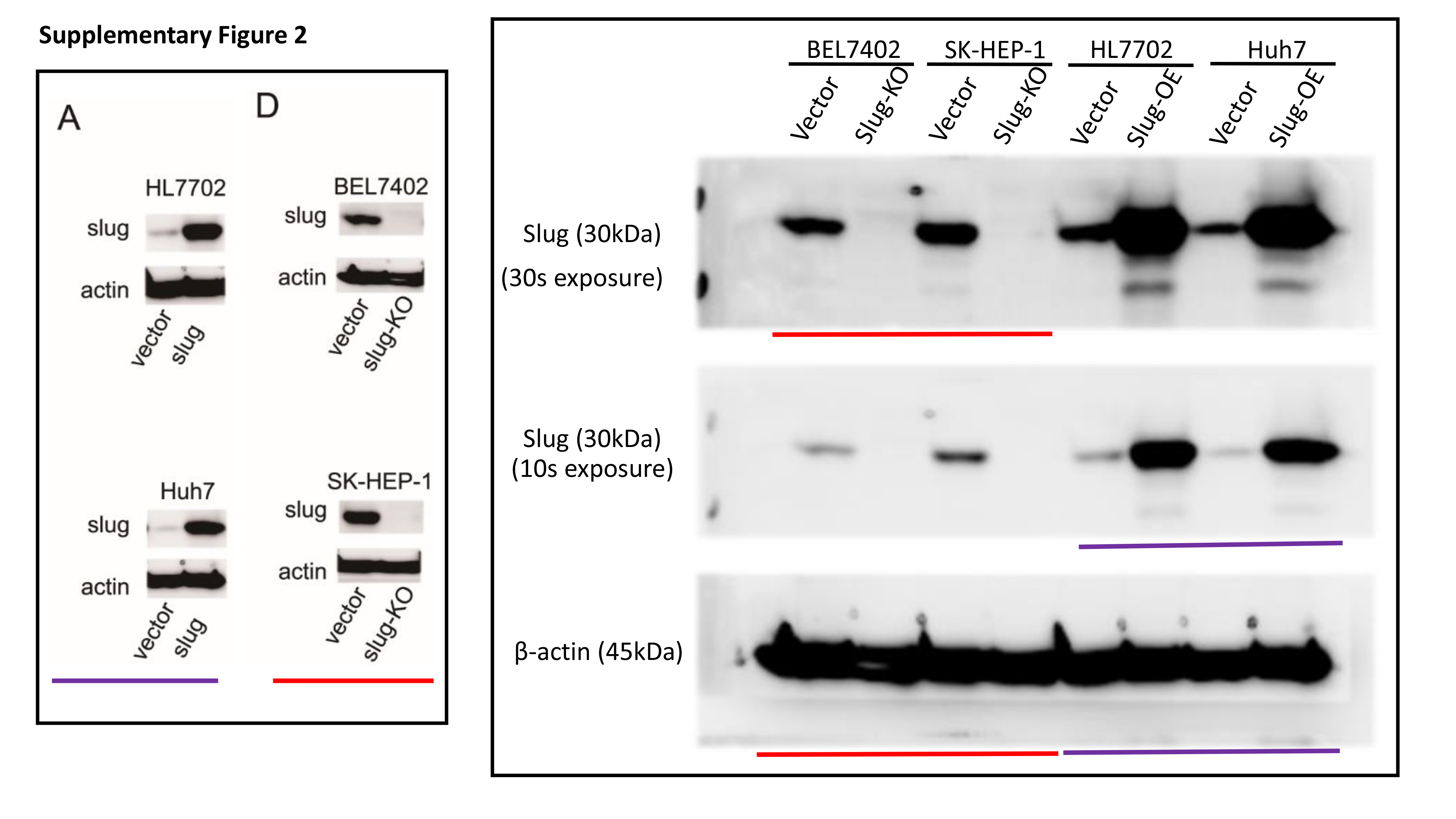
Total protein was extracted from cells with RIPA lysis buffer containing protease inhibitors. The protein concentration of lysates was detected by the Bradford method. 30 μg of protein was separated by electrophoresing on 12% SDS-polyacrylamide gel and blotted onto nitrocellulose filter membranes. The membranes were then blocked with 5% nonfat-milk for 1 hour at room temperature. The blots were then incubated with primary antibodies overnight at 4 °C and horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Immune blots were detected using the ECL detection system. Immunoblotting against β-actin was performed as an internal control.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
A total of 2500 HCC cells were plated in 96-well culture plates in 0.2 mL of medium containing 10% FBS. Then, the cells were incubated for different numbers of days, and 20 μL MTT (5 mg/mL) was added to each well and incubated for 4 hours. The supernatant was removed, and 200 μL DMSO was added to dissolve the viable cell-generated reaction products. The absorbance of well of the plates was recorded using a 96-well microplate reader at a wavelength of 490 nm.
Six-well plate colony formation assay
Five hundred HCC cells were seeded in each well of 6-well culture plates in 2 mL of medium containing 10% FBS. After culture for 2 weeks, cells were fixed in 4% paraformaldehyde and stained with crystal violet. Visible colonies in each well were counted.
Transcriptome analysis (RNA-seq)
Total RNA from each sample was extracted using TRIzol Reagent (Invitrogen). Next generation sequencing libraries were constructed according to the manufacturer’s protocol (NEBNext® Ultra™ RNA Library Prep Kit for Illumina®). Poly(A) mRNA isolation was performed using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB). mRNA fragmentation and priming were performed using NEB Next First-Strand Synthesis Reaction Buffer and NEB Next Random Primers. First-strand cDNA was synthesized using ProtoScript II Reverse Transcriptase, while second-strand cDNA was synthesized with Second-Strand Synthesis Enzyme Mix. The purified double-stranded cDNA (AxyPrep Mag PCR Clean-up [Axygen]) was treated with End Prep Enzyme Mix to repair both ends and to add dA-tailing in one reaction, followed by a T-A ligation to add adaptors to both ends. Size selection of the adaptor-ligated DNA was performed using AxyPrep Mag PCR Clean-up (Axygen), and fragments of ~360 bp (with an approximate insert size of 300 bp) were recovered. Each sample was amplified with polymerase chain reaction (PCR) for 11 cycles using P5 and P7 primers; both primers carried sequences that could anneal with the flow cell to perform bridge PCR. The P7 primer carried a six-base index that allowed multiplexing. The PCR products were cleaned up using AxyPrep Mag PCR Clean-up (Axygen), validated with an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), and quantified using a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA). Libraries with different indices were multiplexed and loaded on an Illumina HiSeq instrument according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Sequencing was carried out using a 2 ´ 150 bp paired-end (PE) configuration; image analysis and base calling were conducted with HiSeq Control Software (HCS) + OLB + GAPipeline-1.6 (Illumina) on the HiSeq instrument. The sequences were processed and analyzed by GENEWIZ Inc. (Hangzhou, China). RNA from each group was extracted and analyzed in triplicate. Fragments per kilobase million (FPKM) was used for statistical analysis. Differentially expressed genes (DEGs) with a p value < 0.05 by Student’s t-test and an average fold change > 1.25 were considered significant.
Real-time quantitative PCR analysis
In brief, 1 μg of total RNA was subjected to reverse transcription using Superscript III transcriptase (Invitrogen, Carlsbad, CA, USA). Real-time quantitative PCR was performed on a Bio-Rad CFX96 system with SYBR Green to determine the mRNA expression level. The reaction conditions used for PCR were as follows: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s. The mRNA expression level was normalized to that of human β-actin. Relative fold changes in mRNA expression were calculated using the formula 2−ΔΔCq [6]. Primer sequences are listed in Supplementary Table 1.
Xenograft animal model
12 four-week-old male BALB/c nude mice weighing 18-23.5 g were obtained from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China) and housed in sterile laminar flow rooms with 12-h light and dark cycles at a temperature range of 19-23˚C and a humidity of 40-60% in the Laboratory Animal Centre of Xi'an Jiaotong University. All experimental procedures were conducted in accordance with the institutional guidelines for conduct and animal welfare. The animals were divided into two groups randomly (six mice for each group). Then they were inoculated subcutaneously into the right dorsal portion with 2×106 Huh7 cells infected with vector or slug overexpression lentivirus. Tumor diameters were measured once a weak. Tumor volumes (V) were calculated with the formula: V=A×B2/2 (A: axial diameter; B: rotational diameter). Mice were killed 4 weeks after injection. Tumor weight was measured, and tumor specimens were analyzed for hematoxylin-eosin (HE) staining and immunohistochemistry staining. Experiments were conducted in accordance with relevant institutional and national guidelines for the care and use of laboratory animals. The mice were raised in 12 hour light/dark cycle at room temperature in a SPF laboratory animal room.
HCC tissues and immunohistochemistry staining Sixty-eight human HCC tissues were collected from patients who underwent partial hepatectomy for HCC at the Department of Hepatobiliary Surgery, the First Affiliated Hospital of Xi’an Jiaotong University, between August 2017 and July 2019. The approval of the institutional review board of the First Affiliated Hospital of Xi’an Jiaotong University was obtained before the samples were collected, which was performed with the permission of the patients. Tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Five-micrometer-thick sections were prepared, and immunohistochemistry staining against slug, p-Akt Ser473 and CDK6 was performed. Immunohistochemistry was performed with the DAKO EnVision™+ System. Evaluation of immunohistochemistry staining was judged by the intensity of staining. Each section was examined under a high-power field (400´) in a double-blinded manner by a pathologist to define the staining of the samples as strong or weak. Clinical and pathological information for HCC tissues is listed in supplementary table 2.
Statistical Analyses
Statistical analyses were carried out using SPSS 16.0 (SPSS Inc., Chicago, IL, USA). Quantitative data are presented as the mean ± SD. Differences among 2 groups were compared using Student′s t test. Analysis of the association between slug and p-Akt or CDK6 expression was carried out using χ2 tests. Significance was assumed for p values < 0.05.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}