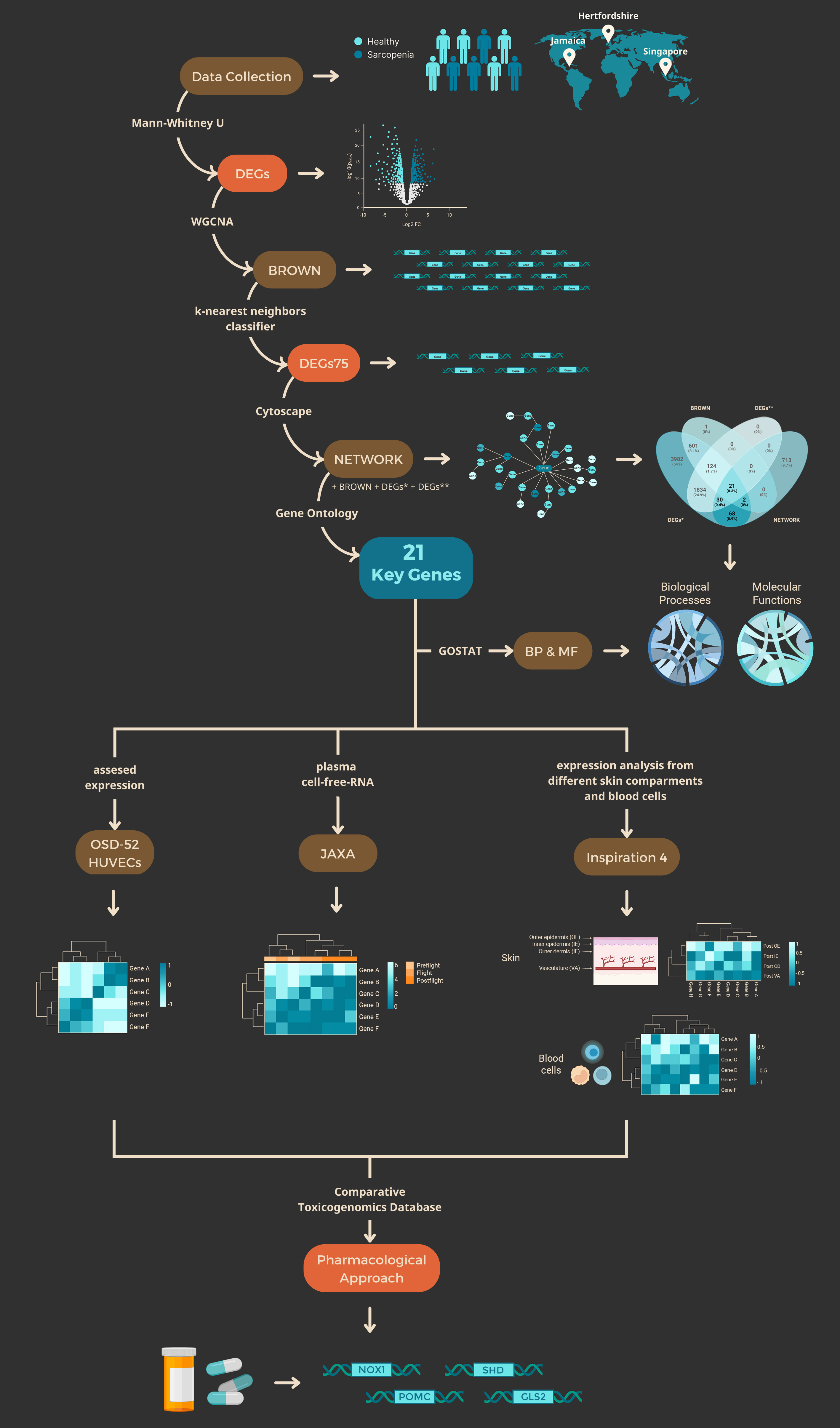
After studying databases from 118 people with and without sarcopenia (GSE111006, GSE111010 and GSE111016) (Migliavacca 2019), we identified 6892 DEGs* through statistical inference and estimated their individual predictive power for that condition by means of a simple classifier, i.e., k-nearest neighbors (Pedregosa et al., 2011).
Then, via co-expression network analysis upon those DEGs*, we identified the module with the highest correlation with sarcopenia, which we denote as BROWN (Fig. 1).
In order to identify linear relationships between DEGs75, we proceed to visualize the resulting Pearson’s correlation coefficients in the form of a heatmap shown in Fig. 2. In this respect, the DEGs75 list contains 62 protein-coding genes, 26 pseudogenes and 23 lncRNAs as the best predictors of sarcopenia. Thus, the ability to predict sarcopenia through the DEGs75 highlights the importance of these non-coding genes as biomarkers.
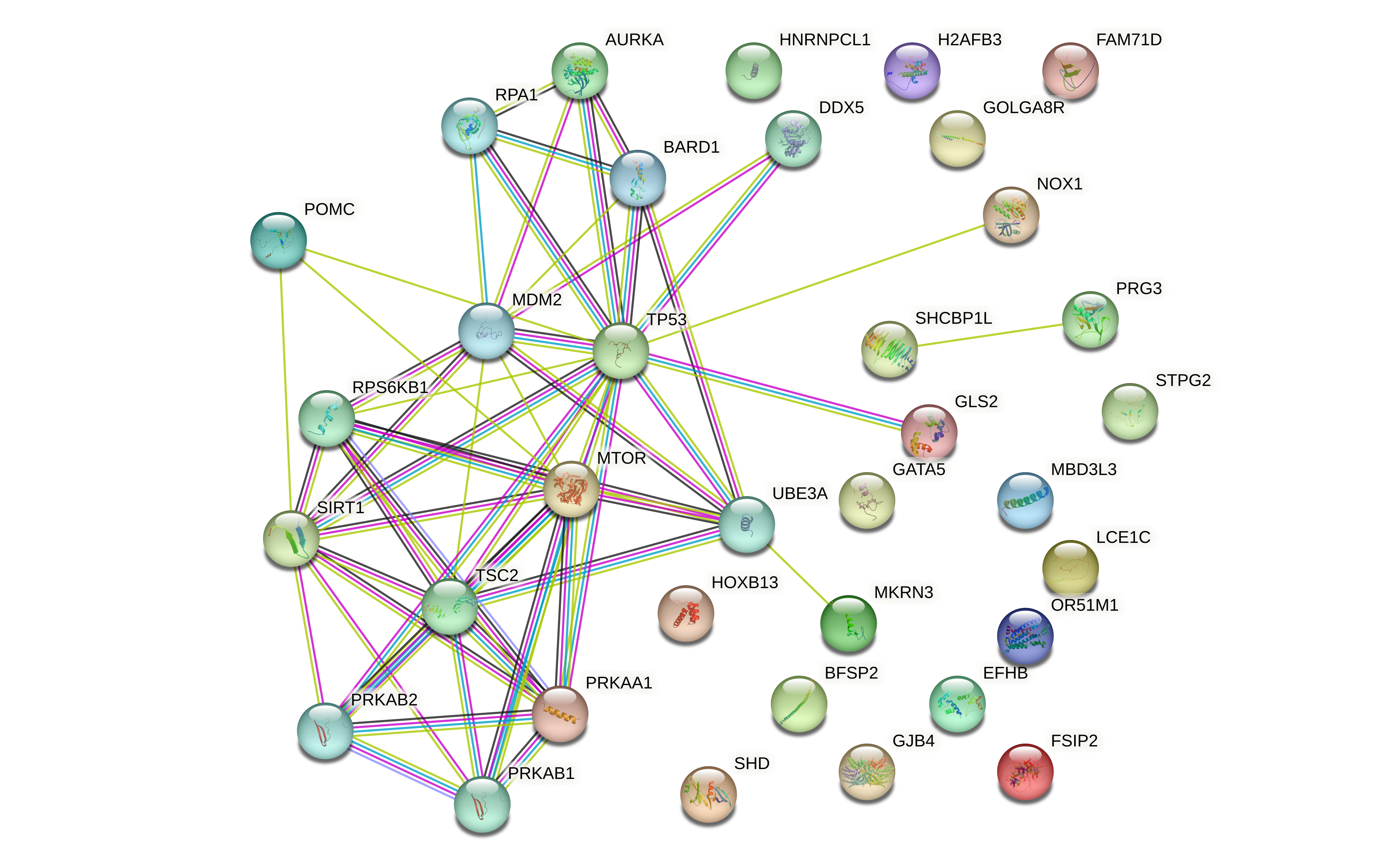
The eigengene from the BROWN module obtained a Pearson correlation coefficient of 0.93 with sarcopenia. Subsequently, we performed pathway and GO analysis upon the 21 genes resulting from the overlapping of BROWN, NETWORK, DEGs* and DEGs** and they all significantly enriched in BP related to sarcopenia, see Figs. 4,5,6. Here, we denote this set of genes as KGs.
The genes in BROWN were mostly downregulated. Only three genes, namely APELA (Apelin Receptor Early Endogenous Ligand), AC023347.2 and NOX1 (NADPH Oxidase 1), had a Fold Change (FC) greater than 1.5, whereas 83 genes had a FC < 0.75. The top downregulated genes were LOC102724594, MT-TQ (Mitochondrially Encoded TRNA-Gln), AC015967.2, AC005014.3, MT-TE (Mitochondrially Encoded TRNA-Glu ) and MFSD6L (Major Facilitator Superfamily Domain Containing 6 Like) with a FC between 0.59 and 0.68.
Additionally, we tested the nutritional gene expression biomarkers currently used to diagnose sarcopenia and found their MAS 19. Not all of the expected genes were differentially expressed, but we found that bone morphogenetic proteins (BMPs) had the highest MAS among the nutritional biomarkers (BMP3 72.28%, BMP6 71.63%, BMP6 71.64% and BMP7 69,41%). Some of the described negative regulators had lower scores such as TGFβ (TGFB1 61.36%), and activins A and B (67.09% and 65.19%) .
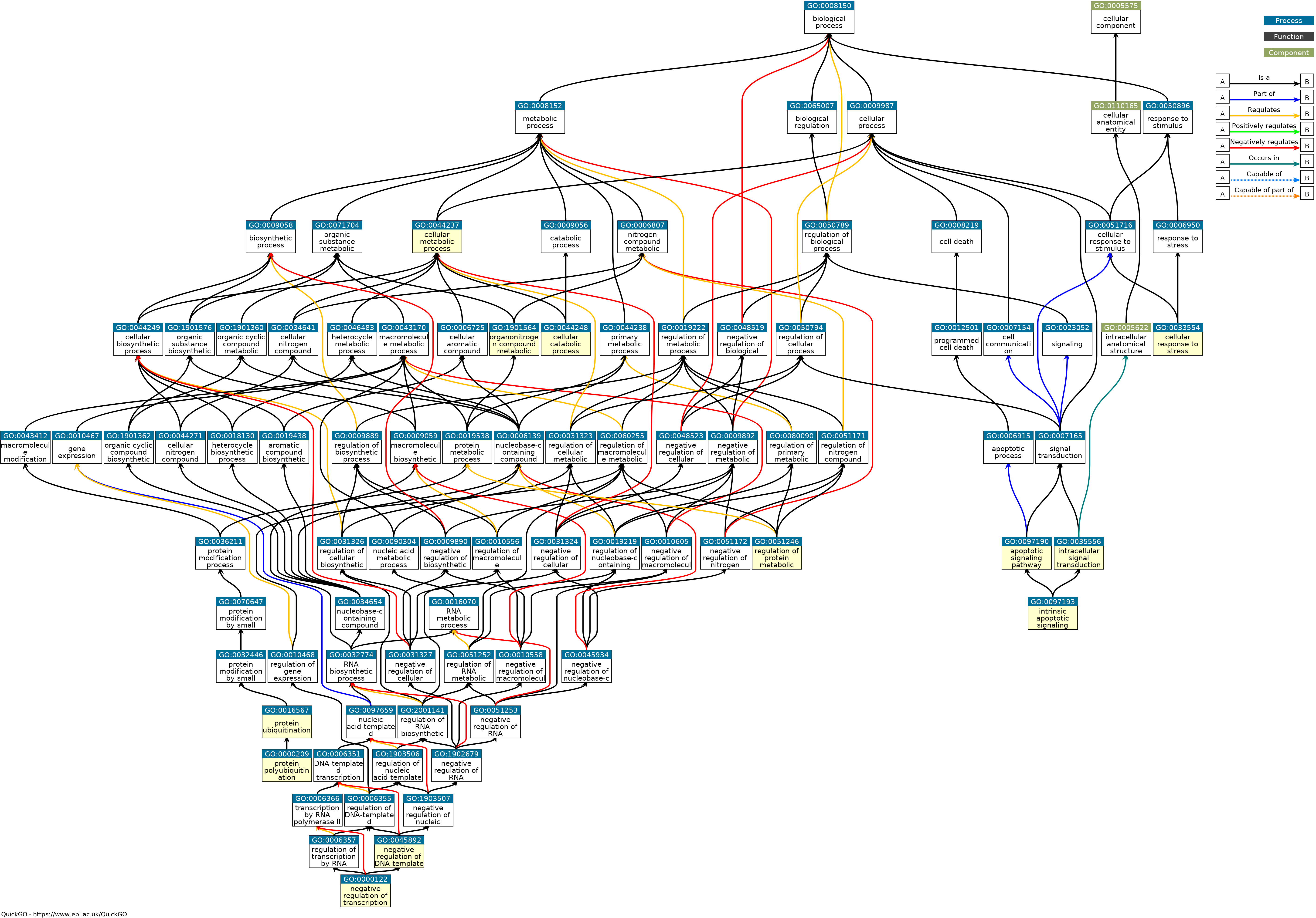
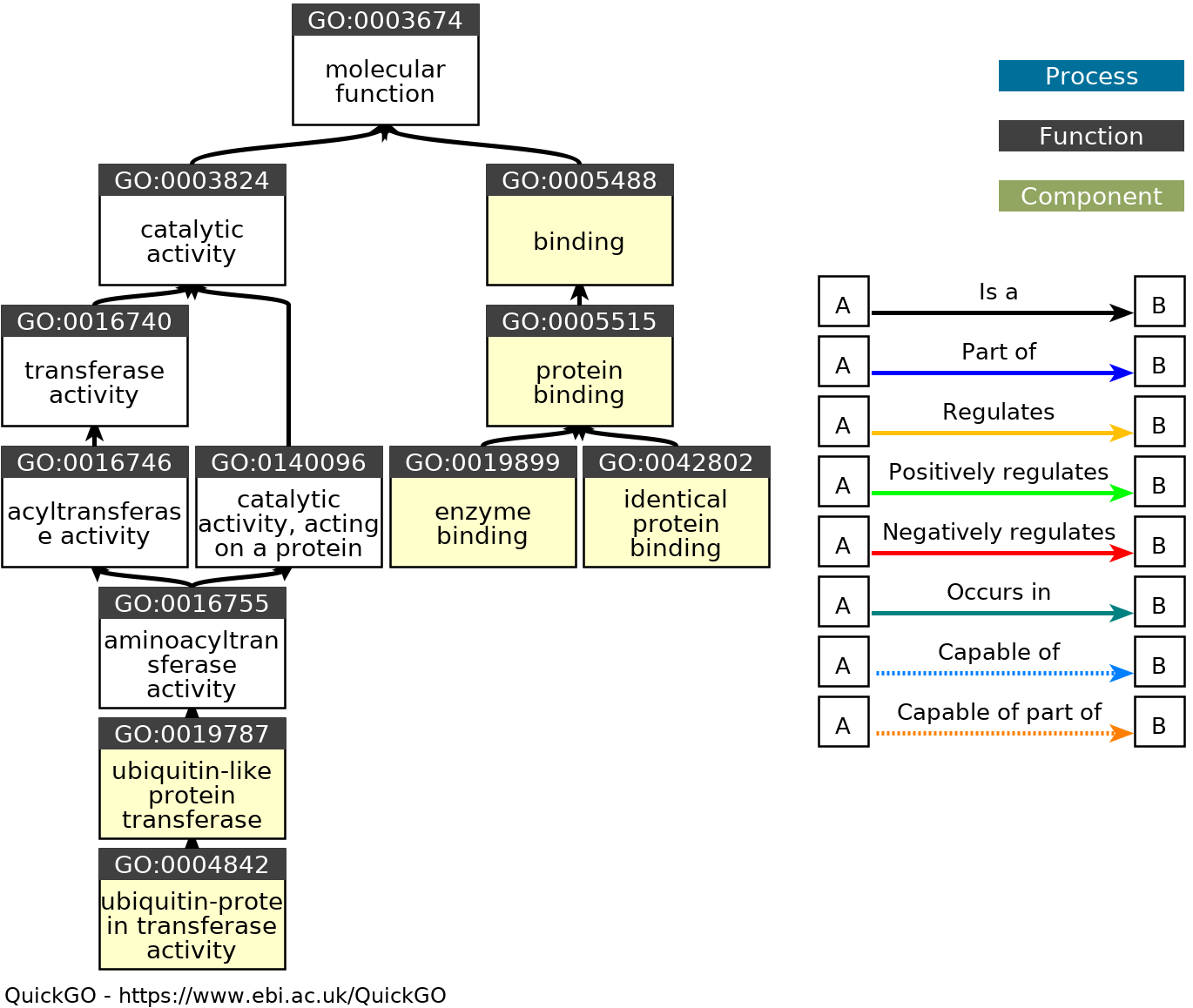
Using the 121 DEGS from NETWORK (NETWORK- DEGs*), we obtained enriched Biological Processes (BP) and Molecular Functions (MF) terms depicted in Fig. 4C and D respectively. Interestingly, when we studied our KGs more closely, we found that five of them (NOX1, MKRN3 (Makorin Ring Finger Protein 3), GATA5 (GATA Binding Protein 5), GLS2 (Glutaminase 2), and POMC (Proopiomelanocortin)) respond to cells' energy levels through TP53 (Tumor Protein P53) and indirectly through MTOR (Mechanistic Target of Rapamycin Kinase), SIRT1 (Sirtuin 1) and PRKAA1 (Protein Kinase AMP-Activated Catalytic Subunit Alpha 1) (Supplementary Fig. 5). In accordance with these results, the bioenergetic alterations could be the result of altered oxidative phosphorylation, mitochondrial energy homeostasis and mTOR signaling related to sarcopenia and muscle loss. On BP we found that the most significant pathways were protein polyubiquitination (GO:0000209) followed by parent terms such as protein ubiquitination (GO:0016567), and protein modification by small protein conjugation or removal (GO:0070647). The next highest enriched pathway was associated with the intrinsic apoptotic signaling pathway (GO:0097193).
Then, to elucidate the role of each KG we searched for their presence in the resulting enriched pathways. NOX1 was present in both of these main processes by being part of all the apoptotic-related pathways and part of a parent GO term to protein ubiquitination. Notably, the gene MKRN3 is directly involved in the ubiquitination of proteins and exclusively related to ubiquitination on our GO result (Fig. 4A). GLS2 and HNRNPCL1 (Heterogeneous Nuclear Ribonucleoprotein C Like 1) were also part of both main pathways, but were only present on the most ancient terms.
The same analysis was performed but now for MF and we found that the most enriched pathways (FDR < .05) were also closely related to protein ubiquitination. More specifically, the ubiquitin conjugating enzyme activity (GO:0061631) was the most enriched pathway. Like in BP, NOX1 and HNRNPCL1 were present in parent terms to the top enriched pathway, but MKRN3 was again present in the child terms (Fig. 4B).
Next, we compared the expression of KGs in both sarcopenic patients and samples from spaceflight. We found that SHD, (Src Homology 2 Domain Containing Transforming Protein D), SHCBP1L (SHC Binding And Spindle Associated 1 Like), MKRN3, GLS2, POMC, EFHB (EF-Hand Domain Family Member B), and STPG2 (Sperm Tail PG-Rich Repeat Containing 2) were downregulated in both spaceflight and sarcopenic conditions, however this change was only significant in sarcopenia (Fig. 4E).
We assessed the expression of our KGs using the OSD-52 dataset obtained from in vitro cultured HUVECs. However, the clustering analysis revealed that the GC replicate 3 had an expression pattern more similar to FLT 1 and 2, while FLT-3 was more similar to GC-1 and 2. Although none of the 21 genes were significantly downregulated, their expression levels tended to be lower (e.g., MKRN3, SHCBP1L, OR51M1 (Olfactory Receptor Family 51 Subfamily M Member 1), and POMC), as shown in Fig. 5. Furthermore, we conducted an analysis using cell-free epigenome RNA-seq data obtained from JAXA, which revealed that the expression of GATA5, HOXB13 (Homeobox B13), SHCBP1L, SHD, GLS2, and MKRN3 tended to increase after spaceflight. It is noteworthy that GATA5, which is known to be increased in patients with sarcopenia, was also found to be elevated in astronauts post-flight. These findings suggest that the in vitro model was affected by spaceflight through different pathways, as the gene expression of the KGs did not appear to be altered. Nonetheless, our results indicate that a cell-free blood sample from astronauts could be used to identify deregulated genes that serve as biomarkers for muscle loss and sarcopenia.
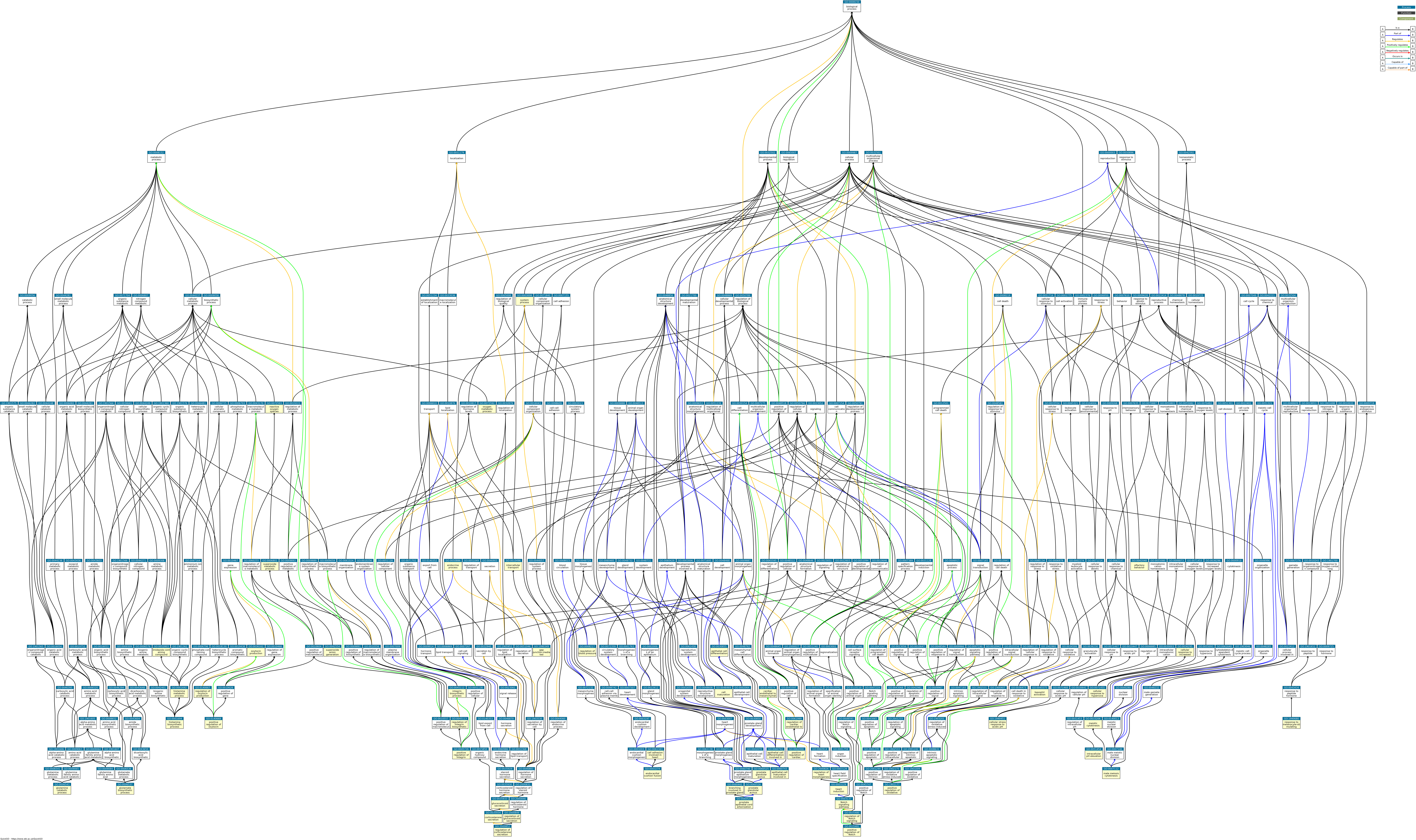
After studying the broad perspective of the NETWORK-DEGs* (Fig. 4), we decided to analyze exclusively the KGs and compared these results to the previous GO enrichment. For BP (Fig. 6) the most relevant terms were related to oxidative stress, pH regulation, epithelial differentiation, integrin biosynthesis, and histamine regulation processes (p. value < 0.05 for BiNGO and p.value < 0.01 for GOstat). We note that there is an enrichment in the cardiac Endothelial to Mesenchymal Transition (EMT) as well as blood pressure regulation, both of which are pathways that involve POMC and NOX1. Additionally, the ROS metabolic process requires the interaction of NOX1, PRG3 and GLS2. These results point to the relevance of oxidative stress, cardiovascular alterations, and possibly a connection between both of these processes.
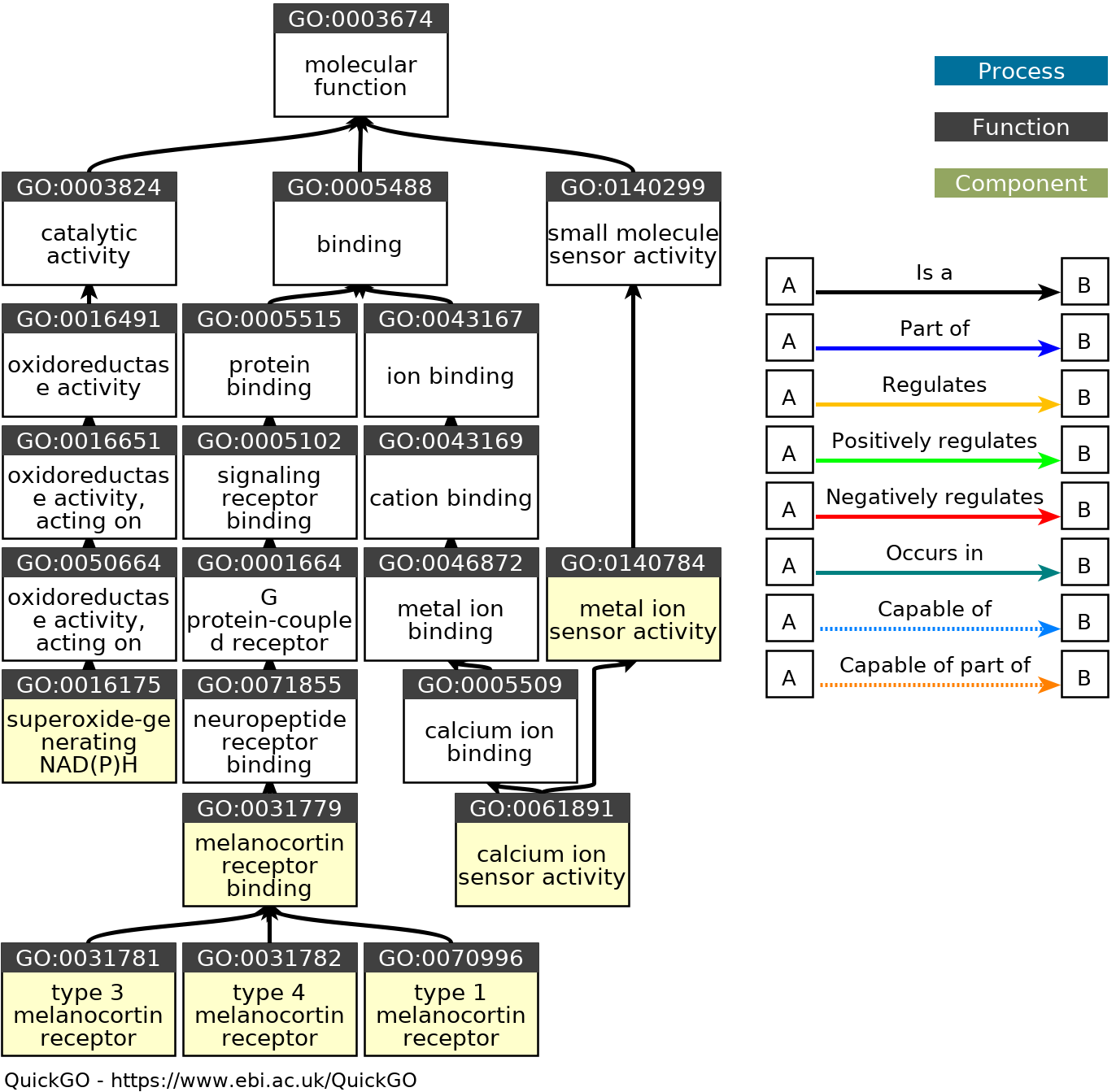
We performed the same analysis for MF and we found an interesting link between POMC and four MF enriched terms (Fig. 7). More specifically, the enriched terms were Superoxide generating NAD(P)H oxidase activity, and melanocortin receptor binding (type I and IV). In contrast to the results from GOstat, through BiNGO we observed that the regulation of hydrogen and proton channels seemed to be enriched. These results are relevant since the hydrogen ion channel activity, oxidoreductase and FAD activity may be related to the mitochondrial metabolism.
During spaceflight, astronauts are exposed to microgravity and other stressful factors that can affect the expression levels of KGs. To investigate these potential changes, we analyzed skin biopsies from the I4 data after spaceflight. We found that 16 out of the 21 KGs showed deregulation, and that the KGs were differentially expressed across the epidermal layers in the I4 data. HOXB13, NOX1, SHD, SHCBP1L, GJB4 (Gap Junction Protein Beta 4), and HNRNPCL1 were overexpressed in two of the four studied compartments (the inner epidermis and outer dermis). Furthermore, six KGs were overexpressed after the flight (OR51M1, STPG2, BFSP2 (Beaded Filament Structural Protein 2), and, to a lesser extent, HNRNPCL1, GJB4, and SHCBP1L) in the vascular compartment. Due to the changes observed in 16 of the KGs in the skin biopsies, this sample type could be informative about the deregulation of genes related to muscle loss in astronauts and the risk of sarcopenia (Fig. 8).
Blood cells from the I4 astronauts could reflect changes in the identified KGs and may represent a less invasive sample source to analyze genes associated with the risk of muscle loss and sarcopenia in astronauts and patients. Blood cells (B-cells, CD4 + and CD8+-Tcells, CD14 + Monocytes, Natural Killers, PBMCs, and other cells) showed different sets of deregulated KGs before and after spaceflight. Out of the 21 KGs, 12 showed significant levels of up and downregulation. The deregulated genes ordered from the most common to the less represented in the samples are: GLS2 (present in 7 blood cell types), STPG2 (6), EFHB (5), MKRN3 (5), BFSP2 (5), FSPI2 (5), GOLGA8AR (5), FAM71D (4), OR51M1 (3), NOX1 (3), SHD (3) and SHCBP1L (2). Interestingly, the majority of the analyzed KGs did not go back to preflight expression levels. In addition, one day after returning to earth, most genes were upregulated representing a possible early stress response.
Based on the fold change values obtained for all KGs in the sarcopenia database, we determined the genes that suffered an increase (i.e., fold change > 1) or decrease (i.e., fold change < 1) in their expression values. We then used this information as a baseline to compare the expression changes upon the same KGs evidenced across data from I4 and JAXA. More specifically, the fold change of each KG in I4 data was computed using the average expression values collected 92 days before flight and 1 day after flight, whereas for JAXA, those expression values were collected 168 days before and 3 days after flight. To be consistent, in both cases we took the most distant measurements taken to the flight date and the closest after flight. Figure 10, shows in green the genes that changed their expression values according to the baseline, and thus in a certain way validate our findings. It is very interesting to see the robustness of POMC, as it behaved identically to our baseline, in terms of down and up regulation, in 9 out of 11 different datasets from JAXA and I4.
Finally, we investigated which KGs could be targeted and regulated by pharmacological agents in clinical trials or already in the market. To begin, we used our KGs to find chemical interactions from the Comparative Toxicogenomics Database (CTD) and found that 4 of them presented data linked to pharmacological interactions. These four genes are POMC, NOX1, GLS2 and SHD. The identified pharmacological agents have already reached at least Phase IV in clinical trials and could therefore be studied for drug repurposing and to possibly target the KGs.
Among the genes studied, POMC seems to be a robust biomarker of sarcopenia, according to Fig. 10, and thus blocking or suppressing its activity with Potassium, Sodium chloride, Lithium, Guanylyl Imidiophosphate and Guanosine Triphosphate could represent a good therapeutic option for people at risk.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}