Reagents and antibodies
Cell culture media, gentamicin, penicillin, and L-glutamine were all obtained from Invitrogen (Grand Island, NY, USA). Tris, NaCl, and SDS for molecular biology and buffer preparation were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies to detect OSGIN1 (15248-1-AP) and GAPDH (HRP-60004) were purchased from Proteintech (Wuhan, Hubei, China). β-Actin (sc-47778), GST (sc-138), His-HRP (sc-8036HRP) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-Flag (F1804) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies to detect Myc (2276), TUBB3 (5568), DYRK1A (8765), MKK3 (8535), MKK6 (8550), p-MKK3/6 (12280), p38 (8690), p-p38 (9211) were purchased from Cell Signaling Technology (Danvers, MA, USA). P-TUBB3 (ab76286) antibodies were purchased from Abcam (Chembridge Science Park, Chembridge, UK).
Construction of expression vectors
Expression constructs, including Myc-OSGIN1, Flag-OSGIN1, Flag-TUBB3 were obtained from GeneCopoeia (USA). Additionally, the lentivirus plasmids shOSGIN1 (#2, 5′- CCGGGGGACAACTTCGTGAGGTTTGCTCGAGCAAACCTCACGAAG
TTGTCCCTTTTTG-3′, #7, 5′-CCGGGGACTTAGACCAGTGTCTGAGCTCGAGC
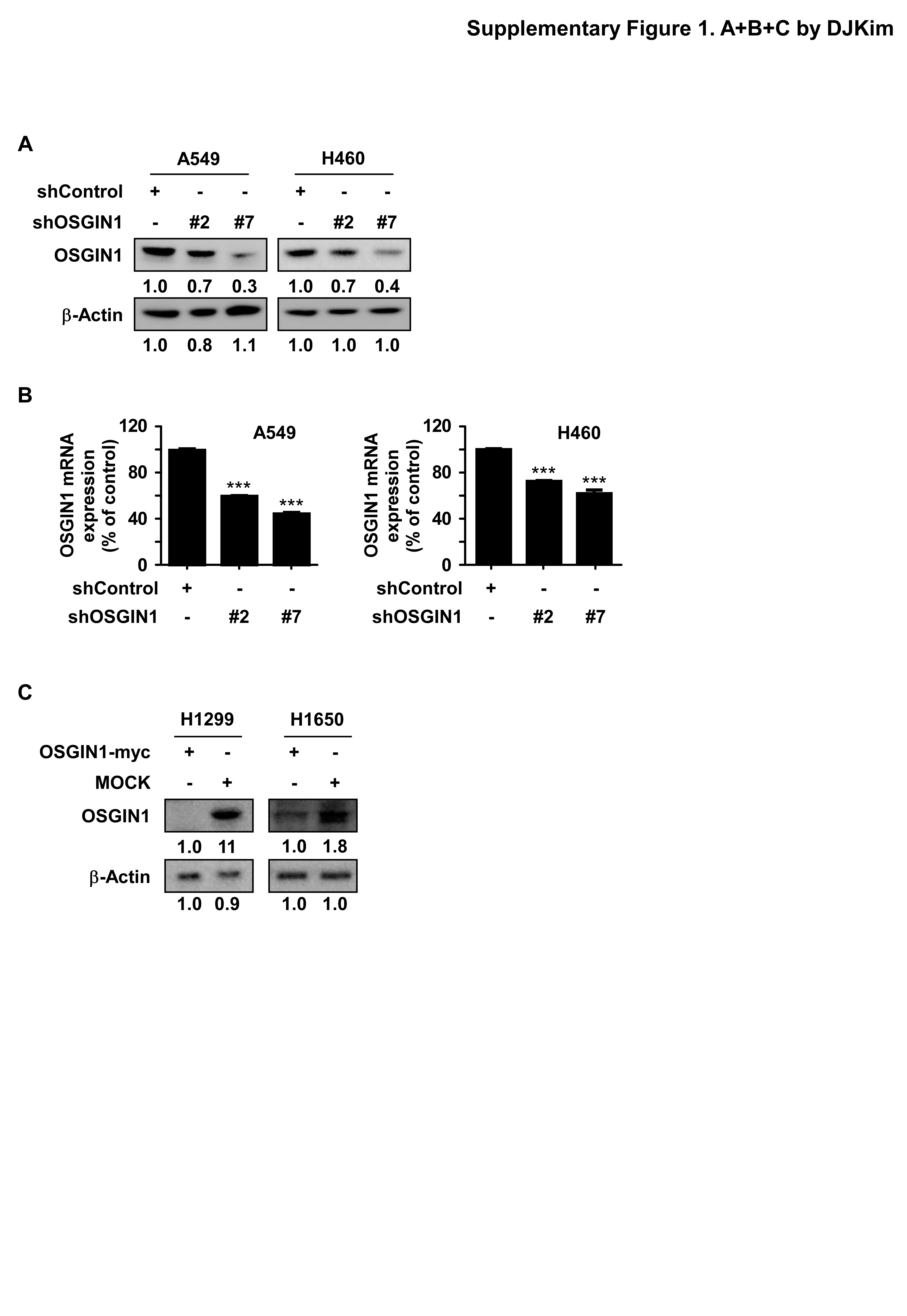
TCAGACACTGGTCTAAGTCCTTTTTG − 3′,) were designed using the Invitrogen BLOCK-iT™ RNAi Designer. The pLKO.1-puro non-target shRNA Control Plasmid DNA (shControl) was purchased from Sigma-Aldrich (St. Louis, MO, USA). All constructs were confirmed by restriction enzyme mapping, DNA sequencing, alignment using the BLAST program.
Cell culture and transfection
Human lung cancer cell lines (A549, H460, H1299, H1650) and the human bronchial epithelial cell NL20 were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Human lung cancer cell lines were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS; Biological Industries, Cromwell, CT, USA) and 1% antibiotic-antimycotic. NL20 cells were cultured in Ham’s F12 supplemented with 0.324 g/L sodium bicarbonate, 0.9 g/L glucose, 1 mM L-glutamine, 0.1 mM non-essential amino acid, 5 µg/ml insulin, 10ng/ml EGF, 1 µg/ml transferrin, 500 ng/ml hydrocortisone, 4% FBS and 1% antibiotic-antimycotic in a 37°C humidified incubator under a 5% CO2 atmosphere. Cells were cytogenetically tested and authenticated before expansion, freezing, and storage in liquid nitrogen. Each cell line was maintained in culture for a maximum of 8 weeks. Transfections were performed using Lipo2000 Transfection Reagent (Invitrogen, Grand Island, NY, USA)) following the manufacturer’s instructions when cells reached 60% confluence. The cells were cultured for 48 h and proteins were extracted for further analysis.
Lentiviral infection
Lentiviral expression vectors of OSGIN1/TUBB3 (shOSGIN1/shTUBB3) or the pLKO.1-puro non-target shRNA Control Plasmid DNA (shControl) and packaging vectors (pMD2.0G and psPAX2) were transfected into Lentix-293T cells using the Lipo2000 transfection reagent following the manufacturer’s instructions. Briefly, the transfection mixture in 10% FBS/DMEM without antibiotics was incubated with cells for 4–6 h. Afterward, the media was discarded and replaced with 10 mL of fresh complete DMEM medium with antibiotics (penicillin/streptomycin). Viral supernatant fractions were collected after 48 h and filtered through a 0.45 µm syringe filter. The filtered virus-enriched media was then supplemented with 10 µg/mL polybrene (Millipore, Billerica, MA) and applied to the target cells. After infection for 48 h, the medium was discarded and replaced with fresh complete growth medium containing the appropriate concentration of 1 µg/mL puromycin. The cells were selected in puromycin for an additional 48 h. The selected cells were used for subsequent experiments.
Cell based assays: cell viability, colony formation, foci formation
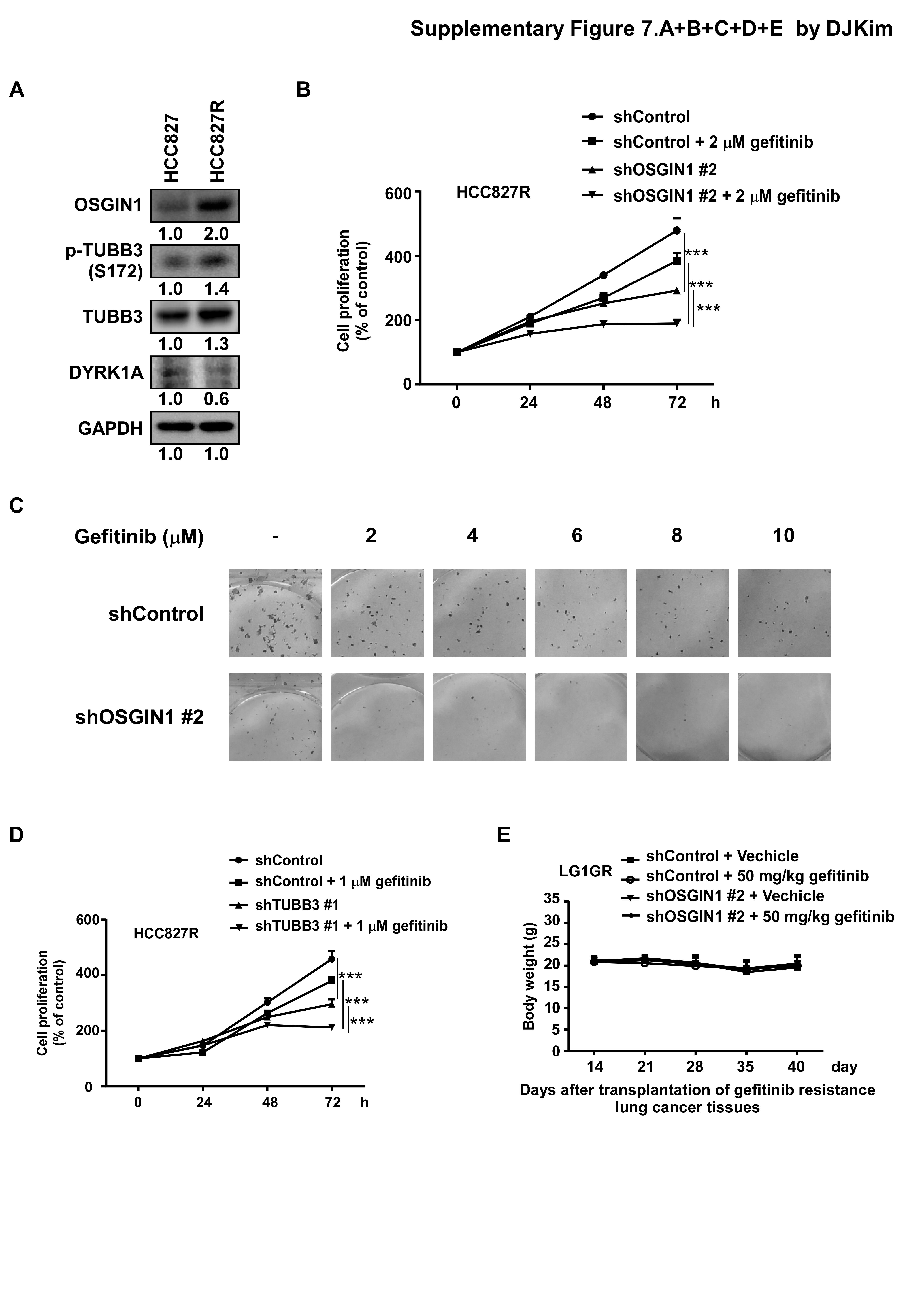
For cell proliferation assays, 2 × 103 cells/well were seeded in 96-well plates and incubated for different time (0, 24, 48 or 72 h) to measure cell proliferation by MTT assay. For anchorage-independent colony formation assays, cells (8 × 103 cells/well) were suspended in complete medium supplemented with 0.3% agar in a top layer over a bottom layer supplemented with 0.6% agar in 6-well plates. The plates were maintained in a 37°C humidified incubator under a 5% CO2 atmosphere for 1 to 2 weeks. For foci formation assays, 500–800 cells/well were seeded in 6-well plates and incubated for 10 to 14 days. The foci were subsequently stained with 0.4% crystal violet and photographed using a camera-mounted wide-field microscope.
Quantitative real-time PCR
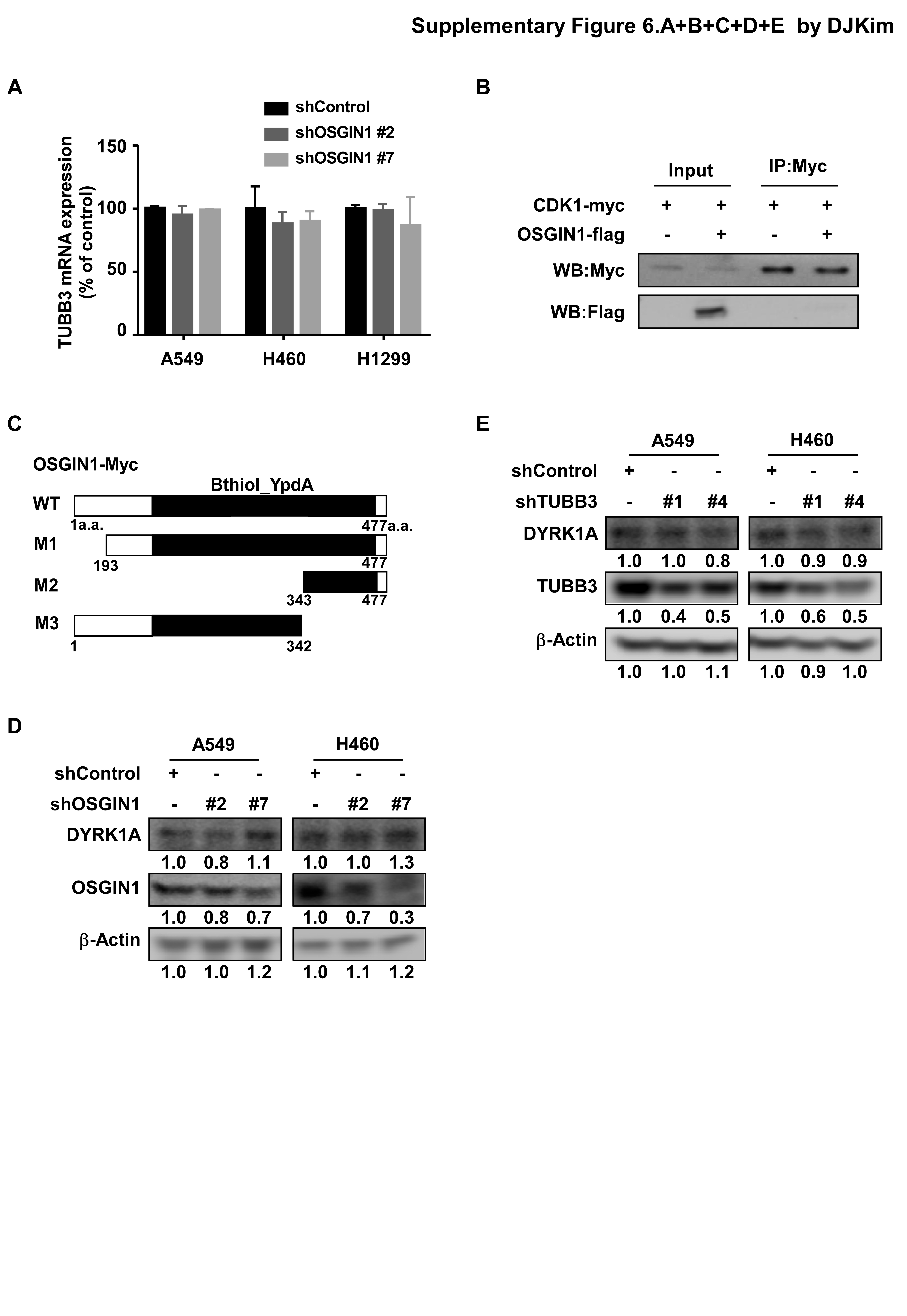
OSGIN1 knockdown and control NSCLC cells (1 × 106) were plated into 100-mm dishes, cultured overnight, and then harvested. An RNA extraction kit (Invitrogen, Grand Island, NY, USA) was used for total RNA extraction. OSGIN1 gene expression was analyzed with 15 ng of total RNA. After cDNA synthesis (Vazyme, Nanjing, China), cDNA was amplified by quantitative one-step real-time PCR following the manufacturer’s suggested protocols. The OSGIN1-specific real-time primers used for RNA quantification are as follows: F:5′- GCCTGGCACTCCATCGAAG − 3′; R:5′- TGACCACGTAGTCCCTGTAGTA − 3′. The TUBB3-specific real-timer primers used for RNA quantification are as follows: F: 5′- GGAGGCACCTCAGACACTCA − 3′; R: 5′- CGATGCCATGCTCATCACTG − 3′. The CT values of OSGIN1 gene expression were normalized with the CT values of actin as an internal control to ensure equal RNA utilization.
Western blotting
Protein concentration were measured by BCA kit (solarbio, Beijing, China) following the manufacturer’s suggested protocol. Proteins were separated by SDS/PAGE and transferred to polyvinylidene difluoride membranes (Amersham Biosciences, Piscataway, NJ, USA). After blocking with 5% nonfat dry milk at room temperature for 1 h, membranes were then incubated overnight with the appropriate primary antibodies at 4°C. The next day, the membranes were washed three times with TBST before and after incubation with 1:10000 dilution of horseradish peroxidase–linked secondary antibody for 1h. The immuno-reactive proteins were detected with chemiluminescence reagent (New Cell ༆ Molecular Biotech, Suzhou, China) using the ImageQuant LA S4000 system (GE Healthcare, Piscataway, NJ, USA).
Tubulin polymerization assay
Lung cancer cells stably expressing shControl or shOSGIN1 were grown in 60-mm plates for 24 h. Afterward, the cells were harvested after washing twice with PBS and then disrupted with 100 µl hypotonic buffer (0.5% NP40, 2 mM EGTA, 1 mM MgCl2, 20 mM Tris-HCl pH 6.8, and a protease inhibitor mixture) for 15 min at room temperature. The lysates were subsequently centrifuged at 13,000 rpm for 15 min at 4°C. After measuring the total protein concentration using a BCA kit (Solarbio, Beijing, China), the soluble fraction containing depolymerized tubulin was separated from the insoluble fraction containing polymerized tubulin. Each fraction was mixed with equal volumes of 5 × SDS loading buffer, heated for 5 min at 95°C, and analyzed by Western blotting.
Surface plasmon resonance (SPR)
SPR assay was performed according to the instructions provided with the Biacore T200 (GE Healthcare, England, UK) instrument. OSGIN1 protein was immobilized onto a CM5 sensor chip. Next, the chip was equilibrated with PBS. A concentration series of TUBB3 protein were added into the flow system to test the binding affinity between TUBB3 and OSGIN1. TUBB3 was dissolved in PBS and perfused onto the CM5 chip at a 30 µl/min flow rate; 120 sec contact time and 300 sec dissociation time were set as the additional parameters. The T200 evaluation state model was utilized to analyze the binding affinity data and calculate the protein’s KD value. Representative curves were re-plotted using the GraphPad Prism software.
In vitro kinase assay
The kinase assay was performed according to the instructions provided by Upstate Biotechnology (Billerica, MA, USA). Active recombinant DYRK1A (100 ng), TUBB3 (300 ng) or OSGIN1 (25, 100 ng) proteins were mixed with ATP and incubated at 30℃ for 30min. The reactions were terminated by adding 5 µl protein loading buffer. Afterward, the mixtures were separated by SDS-PAGE. DYRK1A activity was evaluated using an antibody directed against TUBB3 phosphorylated at serine 172.
Co-immunoprecipitation assay
Cells were co-transfected with OSGIN1-Myc and TUBB3-Flag plasmids. After transfection for 48 h, cell pellets were harvested and incubated with lysis buffer (50 mM Tris-HCl pH 7.4, 1 mM EDTA, 1% TriTonX-100, 150 mM NaCl) supplemented with protease inhibitors for 1 h at 4°C. After quantification, appropriate cell lysates were incubated with beads containing specific tags and rotated overnight at 4°C. The next day, the beads were washed four times with washing buffer (20 mM HEPES pH 7.9, 0.1 M KCl, 0.1 M NaCl, 5 mM EDTA pH 8.0, 0.5% NP40) supplemented with protease inhibitors. The immune complexes were subsequently eluted at 95°C for 5 min with 5× loading buffer. Finally, the immunoprecipitated complexes were visualized by Western blotting.
Immunohistochemistry
The slides containing tissue sections were baked at 65°C for 3 h. After de-paraffinization and hydration, slides were boiled in citrate buffer for 90 sec at a high temperature and pressure. Slides were then treated with H2O2 for 5 min, and incubated with primary antibody at 4°C overnight. Slides were stained with DAB (3, 3'-diaminobenzidine) after incubation with the appropriate secondary antibody. The immunohistochemistry staining was quantified by calculating the integrated optical density (IOD) value measured by Image-Pro Plus analysis.
Immunofluorescence
Cells were grown in a 24-well plate atop 15 mm circle microscope glass coverslips (NEST, USA). After washing the coverslips three times with PBS, samples were fixed in 100% methanol for 15 min. The samples were then blocked with 3% BSA at room temperature for 1 h. Afterward, the coverslips were then incubated with appropriate primary antibodies overnight at 4°C. The next day, the coverslips were washed with PBS before and after incubation with a 1:1000 dilution of the appropriate secondary antibody for 1 h. The cells were then counterstained with with DAPI and mounted onto glass microscope slides. Immunofluorescence images were photographed using a Nikon A1R confocal microscope.
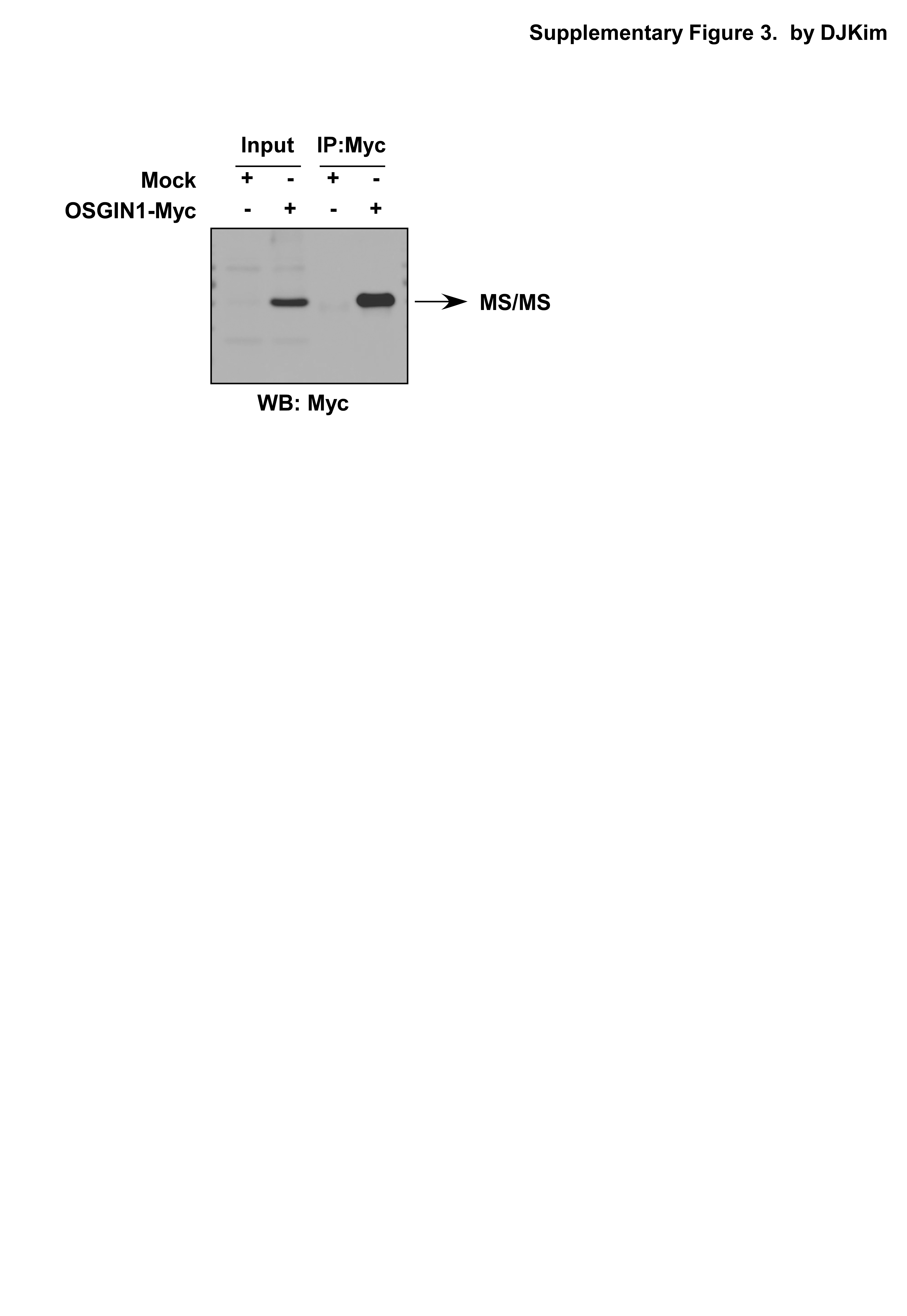
Pull down assay and mass spectrometry
Myc beads were incubated with H1299 lysates (Myc-OSGIN1 transfected only and pcDNA 3.1 vector transfected only as control) for 16 h at 4°C. The samples were then washed four times in washing buffer (20 mM HEPES pH 7.9, 0.1 M KCl, 0.1 M NaCl, 5 mM EDTA pH 8.0, 0.5% NP40) supplemented with protease inhibitors and then subjected to SDS/PAGE. Using CBB staining, discrepant gel lanes were cut down and prepared for mass spectrometry.
Phosphoproteomics
Lysates of cells stably expressing shControl or shOSGIN1 #7 were used for phosphorylated proteomics. The TiO2-enrichment method was used to analyze phosphoproteomics [23].
Computer modeling
The three-dimensional (3D) structures of TUBB3 and DYRK1A are obtained from the Protein Data Bank (PDB Accession Number 6S8L and 2WO6, respectively). DYRK1A is known to phosphorylate TUBB3 at Ser172. Therefore, the DYRK1A catalytic residue Asp287 was specified to be within 8Å to the TUBB3 Ser172 in the docking process. The DYRK1A-TUBB3 model with the best docking score was chosen as the receptor molecule to further dock OSGIN1 onto it. Since OSGIN1 does not have an experimentally defined structure, AlphaFold model (AF-Q9UJX0-F1-model_v4) was used. The final model is consistent with experimental data and has a docking score < -200, which is similar to the values of complex structures in the PDB, indicating a high-confidence model.
Patient-derived lung tumor xenografts (PDX)
Severe combined immunodeficiency (SCID) female mice (6–9 weeks old) (Cyagen, Santa Clara, USA) were maintained under ‘‘specific pathogen-free’’ conditions based on the guidelines established by the Zhengzhou University Institutional Animal Care and Use Committee. Human lung tumor specimens were obtained from the Affiliated Cancer Hospital in Zhengzhou University. Tissues were cut into small pieces and inoculated into the back of the neck of each mouse. Mice were divided into 3 groups consisting of 7–8 animals/group for the HLG77 and HLG80 PDX models as follows: 1) shControl virus infected group; 2) shOSGIN1 #2 virus infected group and 3) shOSGIN1 #7 virus infected group. Previously, an in vivo gefitinib-resistant NSCLC PDX model (LG1GR) was generated in-house [24]. Mice were divided into 4 groups consisting of 7 animals/group for the LG1GR PDX model as follows: 1) shControl virus infected group; 2) shControl virus infected + 50 mg/kg gefitinib group; 3) shOSGIN1 #2 virus infected group and 4) shOSGIN1 #2 virus infected + 50 mg/kg gefitinib group. shControl and shOSGIN1 virus were injected 3 times over the course of 10 days. The gefitinib treatment regimen was initiated when the tumor volume reached approximately 300 mm3. Tumor volume was calculated from measurements of 2 diameters of the individual tumor base using the following formula: tumor volume (mm3) = (length × width × height × 0.52). Mice were monitored until tumors reached approximately 1.5 cm3 total volume, at which time the mice were euthanized and the tumor tissues, liver, kidney and spleen were extracted.
Databases and online survival analysis platform
The expression of TUBB3 in lung adenocarcinoma is obtained from the ualcan website (https://ualcan.path.uab.edu/index.html) [25] and the survival rate is analyzed using Kaplan-Meier Plotter (http://kmplot.com/analysis) [26]. The results of phosphoproteomics was investigated using KEGG (http://www.kegg.jp) [27].
Statistical analysis
All quantitative results were expressed as mean values ± S.D. or ± S.E. Significant differences were compared using the Student’s t test, nonparametric test or one-way analysis of variance (ANOVA). A p value of < 0.05 was considered to be statistically significant. The statistical package for social science (SPSS) for Windows (IBM, Inc. Armonk, NY, USA.) was used to calculate the p value to determine statistical significance.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}