2.1 Mice incisor injury model assay
Experimental animal groups: Seventy Kunming mice were randomly divided into 7 groups: the control group, the abrasion model group, the abrasion LiCl group, the abrasion DKK1 group, the cutting model group, the cutting LiCl group and the cutting DKK1 group. No treatment was done in the control group. The abrasion model group, the abrasion LiCl group and the abrasion DKK1 group used the method of abrasing an incisor. The cutting model group, the cutting LiCl group and the cutting DKK1 group used the method of cutting an incisor.
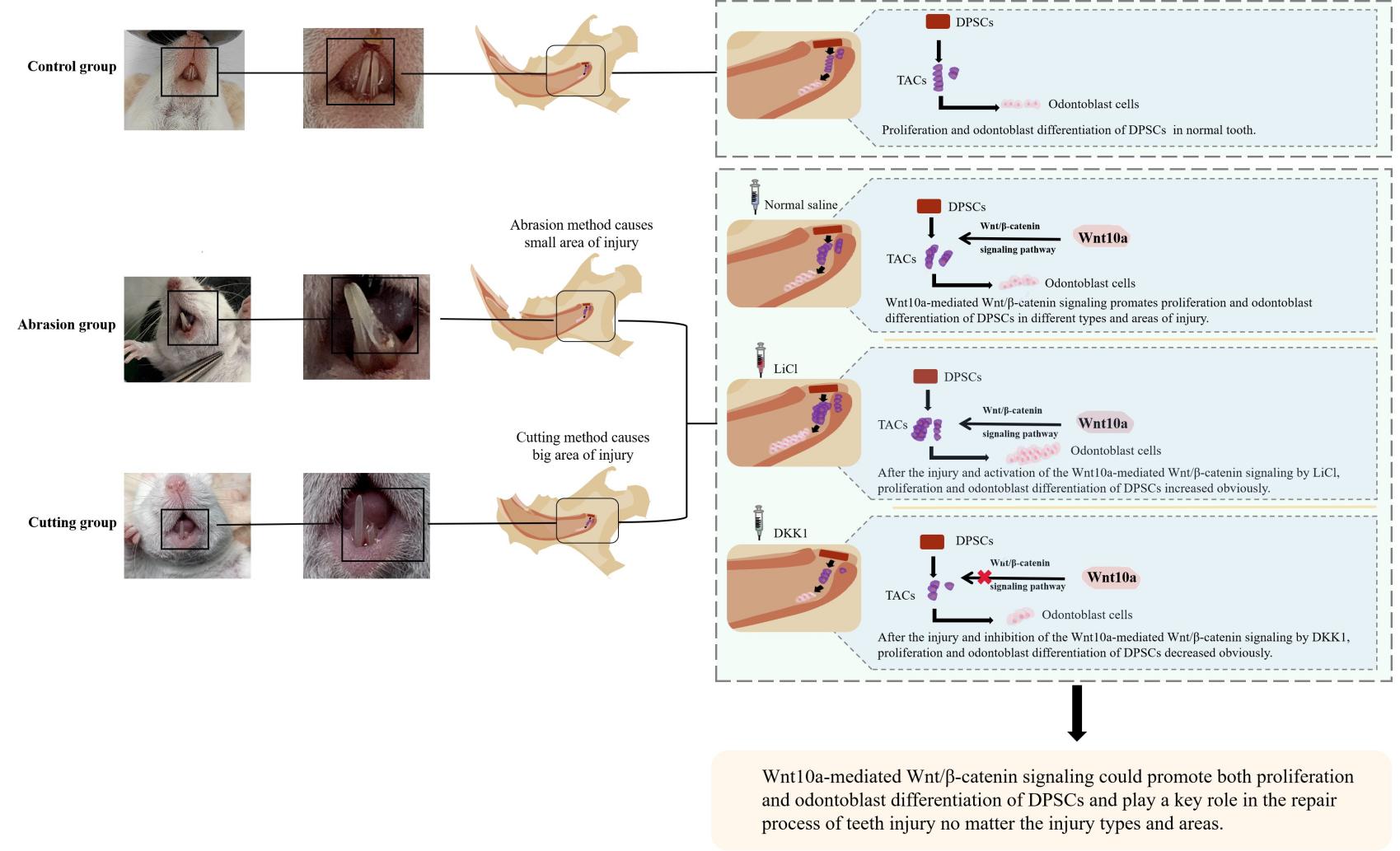
The abrasion method: We used a dental drill to abrade the root of one side incisor, cooling and washing with saline while grinding until the red pulp penetration point was visible to the naked eye. The diameter of the defect was 1.0-1.5mm and the depth was 0.5-1.0mm. The other side incisor was reserved as a negative control. (Fig. 1A1, A1').
The cutting method: We used an ophthalmic scissor to cut directly one side incisor of the mouse until the red pulp penetration point was visible to the naked eye and the other side incisor was still used as a control. (Fig. 1A2, A2').
Drug administration treatment: The abrasion LiCl model group and the cutting LiCl group were given LiCl solution (0.75 g/L) at a dose of 0.2 ml/kg by local injection into the periodontal ligament, once every 2 days. The mice in the abrasion DKK1 group and the cutting DKK1 group were also injected with DKK1 protein at a dose of 0.2 µg/kg into the periodontal ligament, once every 2 days. The cutting model group and the abrasion model group were injected with an equal volume of saline, once every 2 days until the 7th day after the model was constructed successfully. The control group did not receive any treatment.
2.2 qRT-PCR analysis
We extracted an appropriate amount of dental pulp tissue from all groups of mice, added Trizollysis solution to fully lyse, and added chloroform to extract RNA at the ratio of 200ul chloroform per 1 ml of lysis solution. Next, we used the centrifuge at 12000×g, for 15 min at 4℃, took the supernatant, added 0.5 times the volume of isopropanol to precipitate RNA, used the centrifuge at 12,000 × g, for 10 min at 4℃, and discarded the supernatant. In the next step, we added 0.5 ml of 75% ethanol to the precipitate for suspending and washing, used the centrifuge at 7500 rpm for 5 min at 4℃, discarded the supernatant and blotted the mouth of the tube with absorbent paper. Then we opened the lid on the ultra-clean workbench and dried it for 5 minutes in the air. After that, we added 30-50ul of DEPC-treated ddH2O to dissolve the precipitate according to the amount of precipitate, which was total RNA. Referring to the instructions of the SureScript™ First-Strand cDNA Synthesis Kit, we took 1ug of total RNA, added 1ul of SureScriptRTase Mix (20×) and 4ul of SureScript RT Reaction Buffer (5×), replenished water to 20ul and under general conditions (25℃ for 5min, 42℃ for 45min, 85℃ for 5min and 4℃ hold) for reverse transcription on a common PCR machine (4359659, Applied Biosystems 2720 Thermal Cycler, USA) to synthesize the first-strand cDNA. Then we used the BlazeTaq™ SYBR® Green qPCR Mix 2.0 to detect the expression of Axin2 and β-catenin on a CFX96 real-time quantitative PCR instrument (Bio-Rad, USA) by using the Sybrgreen method with cDNA as a template and GAPDH as an internal reference. PCR amplification reaction conditions were: 95℃preheating for 10 min, 95℃ for the 10s, 60℃ for the 20s, 72℃ for the 30s, for 40 cycles, collected and recorded the fluorescence, read the Ct value and used the 2–△△Ct method to calculate the gene relative expression. All the primer sequences required for this study are shown in the table below:
Table 1
The Primer Information Table
| Primers | Sense Primer | Anti-sense Primer |
| Axin2 | TGGACTTCTGGTTTGCTTGTAA | TCTCGTATGTAGGTCTTGGTGG |
| β-catenin | CCTTCACAACCTTTCTCACCAC | ACAGACAGCACTTTCAGCACTC |
| Wnt10a | GGCAGATGGAGGTGTGTGTG | AGGAAGTATGGCCGGGTGTT |
| Dspp | CTCTGTGGCTGTGCCTCTTCTA | CAGTGTTCCCCTGTTCGTTTAC |
| GAPDH | TTGGTCAGGCAAGGGAA | CCCATCACCATCTTCCAGG |
2.3 Immunofluorescence staining
Ki67 immunofluorescence staining: We prepared paraffin sections of 9µm samples at different time points. The paraffin sections were dried at room temperature for 30-50min. Then we washed the sections with 1X phosphate buffered saline (PBS) for 5min, and repeated 3 times. We dissolved TritonTMX-100 in PBS at 1:100 for cell membrane perforations, applied at room temperature for 10 minutes, then washed with PBST for 5min and repeated 3 times. Next, the cells were sealed with Blocking buffer for 1 hour at room temperature and primary antibody Ki67 (1:100) and secondary antibody (1:200) were added sequentially. Subsequently, nuclear staining was performed by using DAPI (1:100), incubated for 5min at room temperature and we protected them from light, washed for 5 min in PBST and repeated 3 times. Finally, fluorescence microscopy was used for image acquisition and analyzed the changes in Ki67-positive cells by Image J image software.
2.4 Western Blotting analysis
Total cellular proteins were extracted using RIPA lysate and proteins were quantified by using the BCA protein assay kit. Samples were electrophoresed through sodium dodecyl sulfate-polyacrylamide gels and transferred topolyvinylidene difluoride membra-nes. Using β-actin as an internal reference control and sealing with 5% fat-free milk, the primary antibody was incubated: Axin2 antibody (1:1000), β-catenin antibody (1:1000), Wnt10a antibody (1:1000), Dspp antibody (1: 2000) and mouse anti-β-actin antibody (1:5000). Then the membrane was incubated with the secondary antibody (goat anti-mouse IgG). Development was performed and exposed. Analyzed protein bands in grayscale were used with Image J software and relative protein expression was calculated.
2.5 Isolation and identification of mouse dental pulp stem cells
Mice incisors were collected and rinsed with PBS containing 5% double antibodies until there was no obvious blood clot on the tooth surface. The pulp in the pulp cavity was removed and rinsed again. Large pieces of pulp tissue were cut up and digested with a mixture of type I collagenase and Dispase II at 37℃. This was followed by culturing with MEM medium containing 20% FBS. The P3-generation DPSCs were identified by flow cytometry after staining by adding 5µl of FITC fluorescently labeled CD44, CD45, CD105, CD34, CD146 and CD31, under light-protected conditions, respectively[15–17, 20].
2.6 Osteogenesis, lipogenesis and chondrogenesis induction and identification of dental pulp stem cells
Osteogenesis induction and identification: P3-genera-tion mouse dental pulp stem cells were inoculated into coated Petri dishes at a cell density of 2×104 cells/cm2, cultured at 37℃ with 5% CO2 to a confluence of 60–70%. Then we discarded liquid supernatant and added osteogenesis-induced differentiation medium (osteogenesis-induced differentiation basal medium 175 ml, specialized fetal bovine serum 20 ml, glutamine 2 ml, dual antibody 2 ml, ascorbic acid 400 µl, β-glycerophosphate sodium 2 mL, dexamethasone 20 µL). About 14 days later, the termination time of cell induction was determined according to the precipitation of intracellular calcium salt crystals and the formation of calcium nodules, and identified by Alizarin Red Staining.
Lipogenesis induction and identification: P3-generation mouse dental pulp stem cells were inoculated, and lipogenesis-induced differentiation mediumsolution A (lipogenesis-induced differentiate-on basal medium A 175 ml, FBS 20 ml, glutamine 2 ml, dual antibodies 2 ml, insulin 400µl, IBMX 200ul, rosiglitazone 200ul, dexamethasone 200µl) and B solution (lipogenic induction differentiation basal medium B 175ml, FBS 20ml, dual antibodies 2ml, glutamine 2ml, insulin 400g) were added to the basal mediums respectively, then the medium was mixed well and placed in the warmed bath. Lipogenic cells were induced by adding the configured lipogenic A solution, changing the B solution once after three days, keeping it for one day and then changing the A solution. Osteogenic induction was added into osteogenic induction mediums, and we placed cells with the added induction medium in the incubator and continued to culture for 14 days. Depending on the condition of the cells, the time to finish the cell induction was decided and Oil Red O Staining was performed for identification.
Chondrogenesis induction and identification: Using the instructions of the chondrogenesis induction kit, P3-generation mouse dental pulp stem cells were digested and counted at 3×105cells in 15 ml centrifuge tubes, respectively, and centrifuged at 220 g for 8 min at low speed to allow the cells to form microclusters. The cells were cultured in centrifuge tubes with chondrogenic induction solution, and we changed liquid at 3–4 d intervals, induced for 5 weeks, fixed the cells in 4% paraformaldehyde and paraffin sections were made routinely. The sections were dewaxed in xylene, dehydrated in gradient ethanol, and washed in distilled water before being stained with Alcinium Stain for identification.
2.7 Effect of LiCl and DKK1 treatment on the proliferation of mouse dental pulp stem cells
The P3-generation dental pulp stem cells were digested, supernatant liquid discarded by centrifugation, washed twice in PBS buffer, counted and transferred to a 6-well plate with the corresponding volume of cell suspension and then incubated with 0.75 g/L LiCl and 1ug/ml DKK1 for 24 h. The proliferation of cells in different groups was detected by EDU staining.
2.8 Statistical analysis
In this study, SPSS 19.0 software was used to statistically analyze the data and the measurement data were expressed as (x ̅ ± s). Independent sample t-test was used for inter-group comparison, and paired t-test was used for intra-group comparison. Enumeration data were expressed as rate (%) and compared by chi-square test. P < 0.05 was considered statistically significant.
{kind=link}