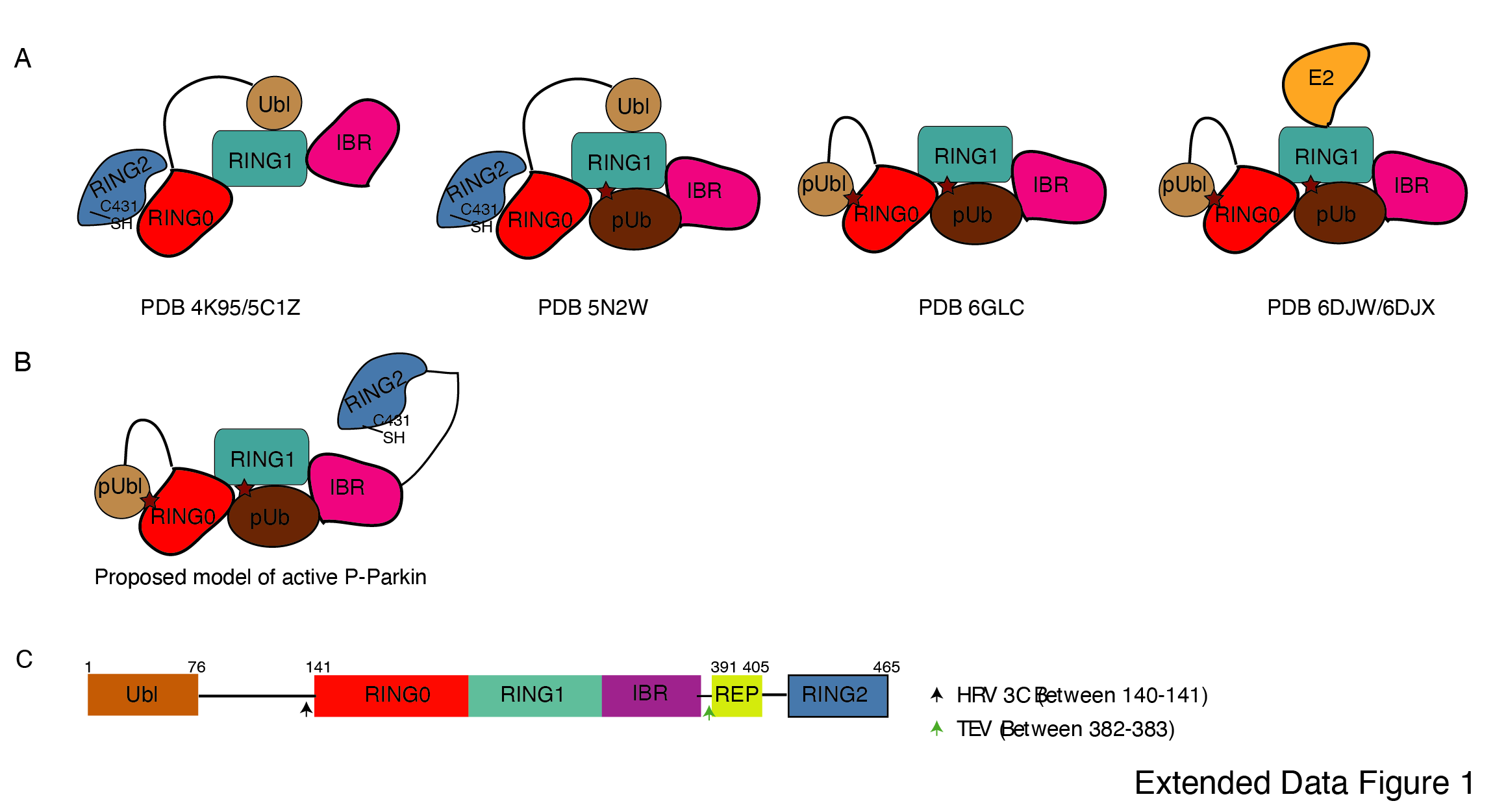
Incorporation of molecular scissors to capture intricate dynamic conformations on Parkin
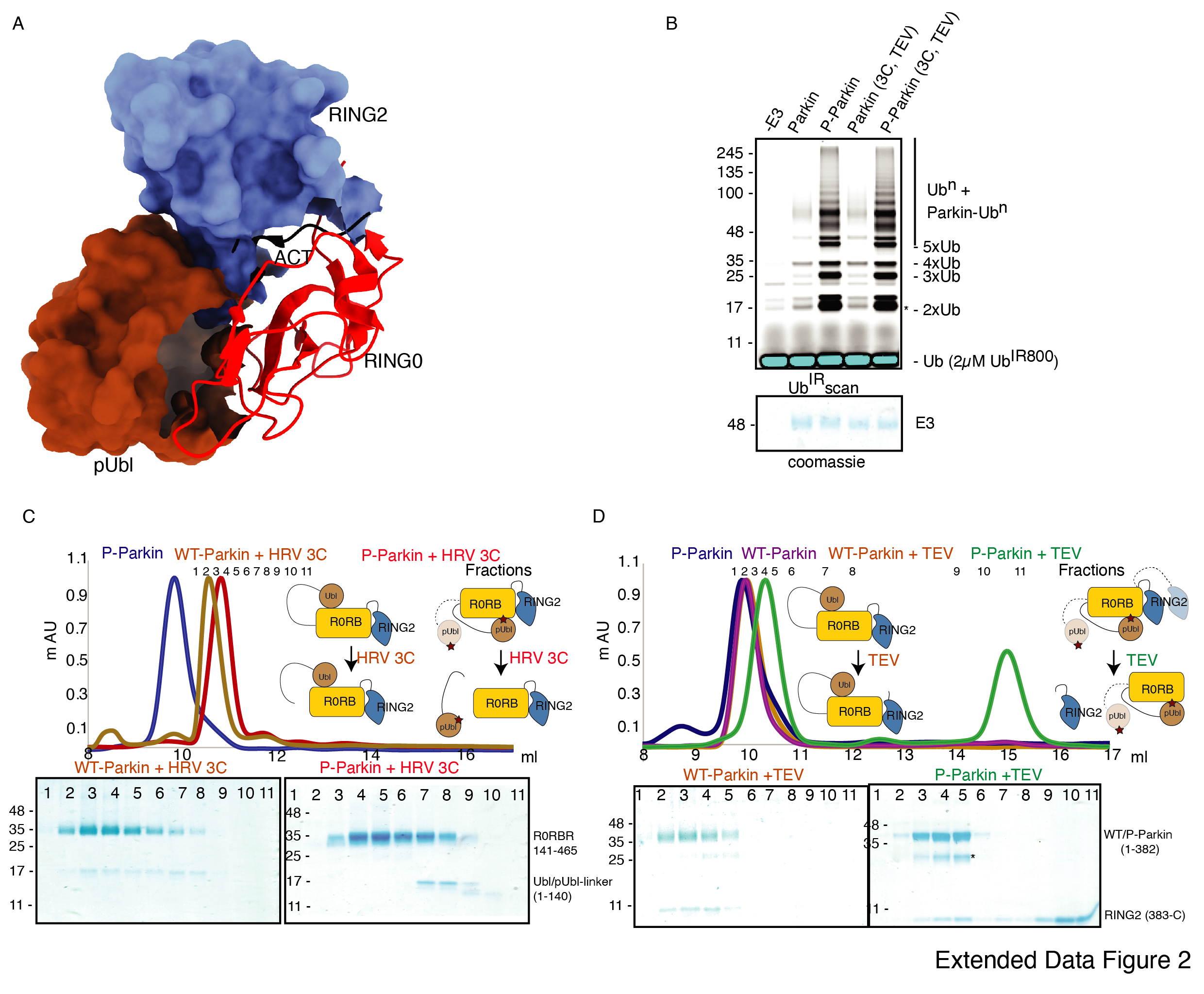
Previous studies using various biophysical methods showed that after phosphorylation of the Ubl domain of Parkin, phospho-Ubl does not interact with the core of Parkin, lacking the Ubl domain 25–27. However, the crystal structure of RING2 removed phospho-Parkin (1-382) showed phosphorylated Ubl domain binds with the basic patch (Lys161, Arg163, Lys211) on the RING0 domain 24, 29. RING2 shares a large surface with RING0, and the superposition of phospho-Parkin (1-382, PDB 6GLC) and WT-Parkin (PDB 5C1Z) structures show steric clashes between RING2, ACT, and pUbl (Extended Data Fig. 2A). Therefore, we wondered whether the RING2 domain competes with the pUbl domain and thus blocks the interaction of pUbl with RING0. The latter hypothesis would also explain why previous attempts to study pUbl interactions show weak or no interactions between pUbl and Parkin in trans.
To capture crystal structures of protein-protein complexes, researchers use fusion construct to allow the expression of two proteins in a single polypeptide chain. The fusion method increases the effective net concentration of two proteins in solution compared to mixing two proteins separately, thus stabilizing the interactions between two proteins. Earlier binding assays on Parkin failed to capture interactions in trans, and we speculated that this might be due to the lower net concentration of the domain in trans compared to the high net concentration of fused domain. We hypothesized that untethering (cleavage of peptide bond) upon protease treatment would solve the above problem and enable us to capture the binding in trans using biophysical methods. To understand the above intricate mechanism, we introduced molecular scissors (human rhinovirus type 3C (HRV 3C) and tobacco etch virus (TEV) on full-length Parkin to analyze the Ubl and RING2 domain rearrangements under native or phosphorylated states. We introduced HRV 3C (between 140th -141st residue) or TEV (382nd -383rd) sites in the loop regions of Parkin (Extended Data Fig. 1C) to avoid any artifacts due to perturbations in native interactions on protein.
First, we tested the ubiquitination activity of the Parkin construct with HRV 3C and TEV sites to ensure that the inclusion of these sites did not affect the protein folding or function, which is confirmed by the similar activity of this construct as of the native Parkin construct (Extended Data Fig. 2B). Furthermore, we noticed that Ubl-linker (1-140) co-elute with R0RBR (141-465) in native Parkin after treatment with 3C protease, suggesting a stronger interaction between Ubl and the Parkin core (Extended Data Fig. 2C). However, when phosphorylated Parkin was treated with 3C, pUbl-linker (1-140) did not form a complex with R0RBR (141-465), suggesting a poor/no interaction between phospho-Ubl with the core of Parkin (Extended Data Fig. 2C). Furthermore, when we treated native Parkin with TEV, RING2 (383-465) co-elute with Parkin (1-382), suggesting a stronger interaction between RING2 and the Parkin core in the native Parkin (Extended Data Fig. 2D). However, when we treated phosphorylated Parkin with TEV, RING2 (383-465) elute separately from the Parkin (1-382), suggesting that phosphorylation of the Ubl domain results in the displacement of the RING2 domain (Extended Data Fig. 2D). All the above data confirmed that inclusion of molecular scissors on Parkin constructs do not affect Parkin folding and the previous observations, that phosphorylation of Ubl weakens its interaction with Parkin core and leads to displacement of RING2, are confirmed using our assay. Also, respective proteases only cleaved (untethered) the peptide bond without affecting the native interactions between Parkin domains.
The phospho-Ubl domain and RING2 domain have a competitive mode of binding on the RING0 domain
We wanted to test whether RING2 and pUbl affect the binding of each other on Parkin, which would suggest a competitive binding mode between pUbl and RING2 on the RING0 domain, or as suggested previously, pUbl negatively affects RING2 binding and lead to RING2 displacement permanently. To test the competitive mode of binding between pUbl and RING2 on RING0, and thus affecting the binding of each other, we performed the SEC assay after sequential treatment with HRV 3C and TEV on Parkin construct with both protease sites as shown in Extended Data Fig. 1C. Interestingly, pUbl-linker (1-140) co-elute with Parkin core (141-382) upon 3C treatment on fractions that were collected after TEV treatment on phospho-Parkin which lead to displacement of RING2 (383-465) (Fig. 1A). Similarly, RING2 (383-465) co-elute with Parkin core (141-382) upon TEV treatment on fractions that were collected after 3C treatment on phospho-Parkin, which led to displacement of pUbl-linker (1-140) (Fig. 1B). This data confirms that pUbl and RING2 competitively bind on RING0. The binding of one negatively affects the binding of the other, unlike previous observations, which only showed phosphorylation of Ubl leading to RING2 displacement.
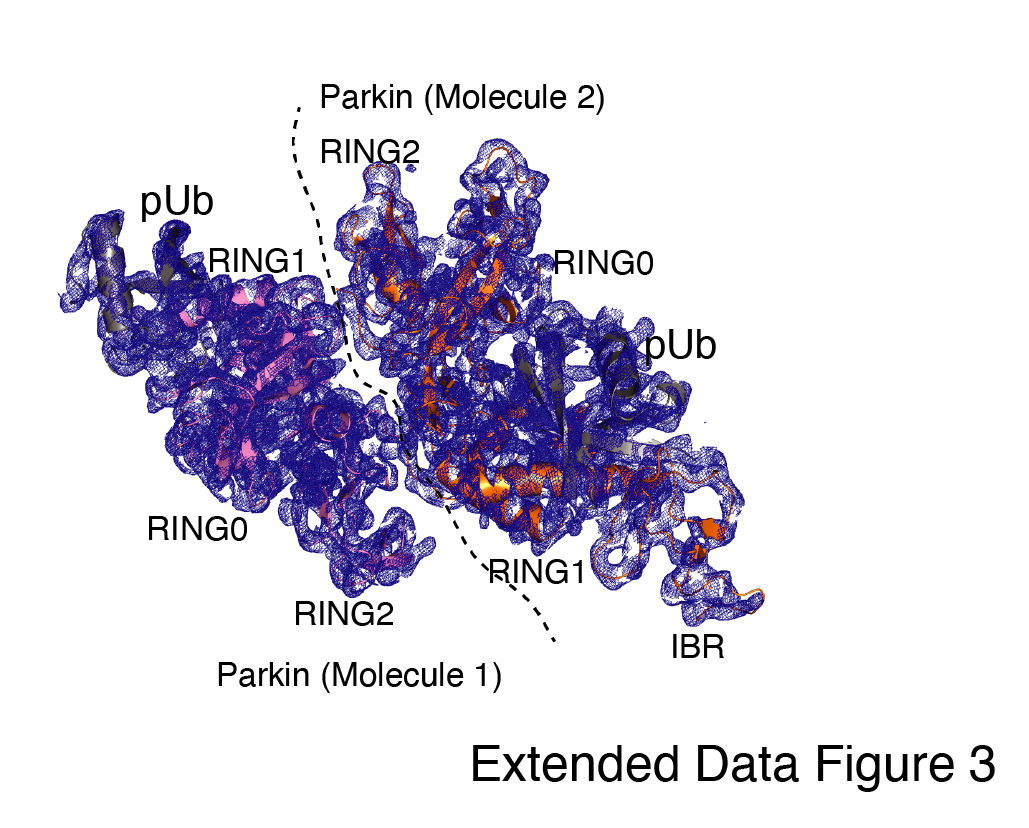
Furthermore, we wanted to test whether RING2 is permanently displaced from RING0 after the pUbl binding on the RING0 domain. To test this hypothesis, we used phosphorylated Parkin, which had both TEV and 3C sites. We crystallized the phospho-Parkin complex with phospho-ubiquitin after treatment with 3C protease, which resulted in the washing off of pUbl-linker (1-140) from Parkin (141-465). The overall structure of pUbl-linker (1-140) depleted Parkin (141-465) in complex with pUb was determined at 3.3 Å (Extended Data Table 1), and showed similar conformation as seen in previously solved Parkin structures in their autoinhibited state (Fig. 1C). The crystal structure shows RING2 bound with RING0, which confirms that RING2 is transiently displaced from the RING0 domain and returns to its original position after washing off pUbl-linker (Fig. 1C), further confirming our SEC data (Fig. 1B). The crystal structure also revealed that the REP element is bound with the RING1, similar to the Parkin structures captured in the autoinhibited state (Fig. 1C). The phospho-ubiquitin was seen bound in the basic patch between RING0 and RING1 and resulted in similar conformational changes in IBR and helix between RING1-IBR domains (Fig. 1C). In the crystal structure, two molecules of Parkin bound with pUb were seen; however, in one of the Parkin molecules, no density was observed in the IBR region (Extended Data Fig. 3). Overall, this data suggest that pUbl and RING2 exist in a dynamic state in phospho-Parkin (pUbl binding<->RING2 open<->pUbl displaced<->RING2 closed) and compete for binding on RING0, unlike previous studies suggesting that only the open conformation of Parkin is mediated by RING2 displacement by pUbl.
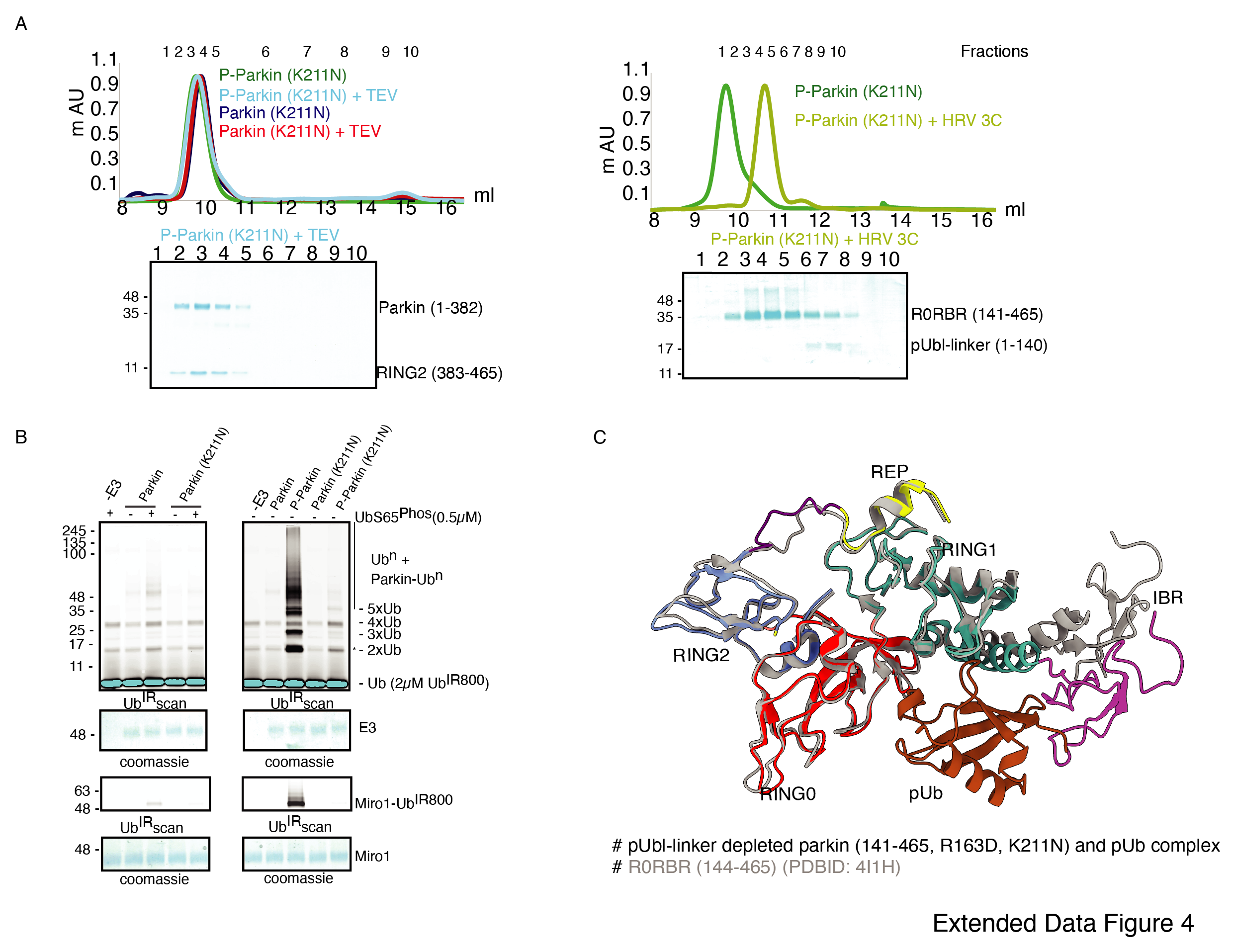
Interestingly, similar to phospho-Parkin (Extended Data Fig. 2C), pUbl-linker (1-140) remained flexible in phospho-Parkin (Lys211Asn) and eluted separately from Parkin core (141-465) on SEC (Extended Data Fig. 4A), suggesting binding with the basic patch on RING0 domain may not be the driving force for pUbl displacement. Further, to confirm that displacement of the RING2 domain is mediated by pUbl binding in the basic patch (Lys161, Arg163, and Lys211) on the RING0 domain, we tested the RING2 displacement using Lys211Asn mutant of Parkin. Mutation of Lys211Asn resulted in stabilization of RING2 (383-465) domain on phospho-Parkin (1-382) upon TEV treatment, and the two fragments co-eluted on SEC (Extended Data Fig. 4A). Although pUbl was displaced in phospho-Parkin (Lys211Asn), Lys211Asn mutation reduced Parkin activity drastically (Extended Data Fig. 4B), suggesting RING2 displacement, not Ubl displacement, is a major cause of Parkin activation. Also, we noticed a basal level of Parkin activity in the lanes without any activator, which was reduced in Lys211Asn Parkin (Extended Data Fig. 4B). To understand the conformational changes upon mutation in the basic patch on RING0, we also crystallized phospho-Parkin (Arg163Asp, Lys211Asn, HRV 3C site at 141st position) in complex with phospho-ubiquitin after treatment with 3C protease, which resulted in washing off of pUbl-linker (1-140) from Parkin core (141-465). This complex resulted in better crystals diffracting up to 2.35 Å. The overall structure of the pUbl-linker (1-140) depleted Parkin (141-465, Arg163Asp, Lys211Asn) in complex with pUb is similar to the autoinhibited structure wherein RING2 is bound on RING0 and REP element is bound on RING1 (Extended Data Fig. 4C).
Untethering of the linker connecting IBR and RING2 allows pUbl binding in trans
We wondered whether the competing nature of binding between pUbl and RING2 could be the reason behind no interaction seen previously between pUbl and Parkin (lacking Ubl domain) in trans 19-30. To test this, we used phospho-Parkin (Lys211Asn), which would not allow the binding of pUbl in the RING0 pocket of the same molecule, and tested its interaction with ∆Ubl-Parkin. However, no complex formation was seen on SEC (Fig. 2A). SEC assay was validated using ITC assay which did not show any detectable interaction between phospho-Parkin (Lys211Asn) and ∆Ubl-Parkin (Fig. 2A). This trans experiment is consistent with our cis experiment (Fig. 1). As our data suggested that the fused domain outcompete the untethered domain (Fig 1), we wondered whether the same could be the reason behind the lack of detectable binding in trans. To test this, we used acceptor Parkin with an untethered linker (TEV site between 382nd - 383rd), which overcomes the problem of higher net concentration of the fused competing RING2 domain. Acceptor ∆Ubl-Parkin (TEV site between 382nd - 383rd) was treated with TEV, and TEV was removed using an affinity column followed by SEC. SEC showed co-elution of ∆Ubl-Parkin (77-382) and RING2 (383-465), confirming that TEV cleaved (untethered) the peptide bond (connecting IBR and REP-RING2) without affecting the native interactions between ∆Ubl-Parkin (77-382) and RING2 (383-465) (Fig 2B). Incubation of phospho-Parkin (Lys211Asn) with untethered ∆Ubl-Parkin led to a stable trans complex on the SEC, showing co-elution of phospho-Parkin (Lys211Asn) and ∆Ubl-Parkin (77-382), leading to the displacement of RING2 (383-465) from ∆Ubl-Parkin (Fig. 2B). The ITC assay showed a strong affinity (Kd = 1.3 ± 0.2 µM) between phospho-Parkin (Lys211Asn) and untethered ∆Ubl-Parkin (Fig. 2B), which further supports SEC assay. We noticed protein precipitation in the cell during ITC measurements, which explains the poor stoichiometry due to protein destabilization after pUbl binding and RING2 displacement. A significant decrease of 3⁰C in the melting temperature of phospho-Parkin 24 is consistent with the above observation.
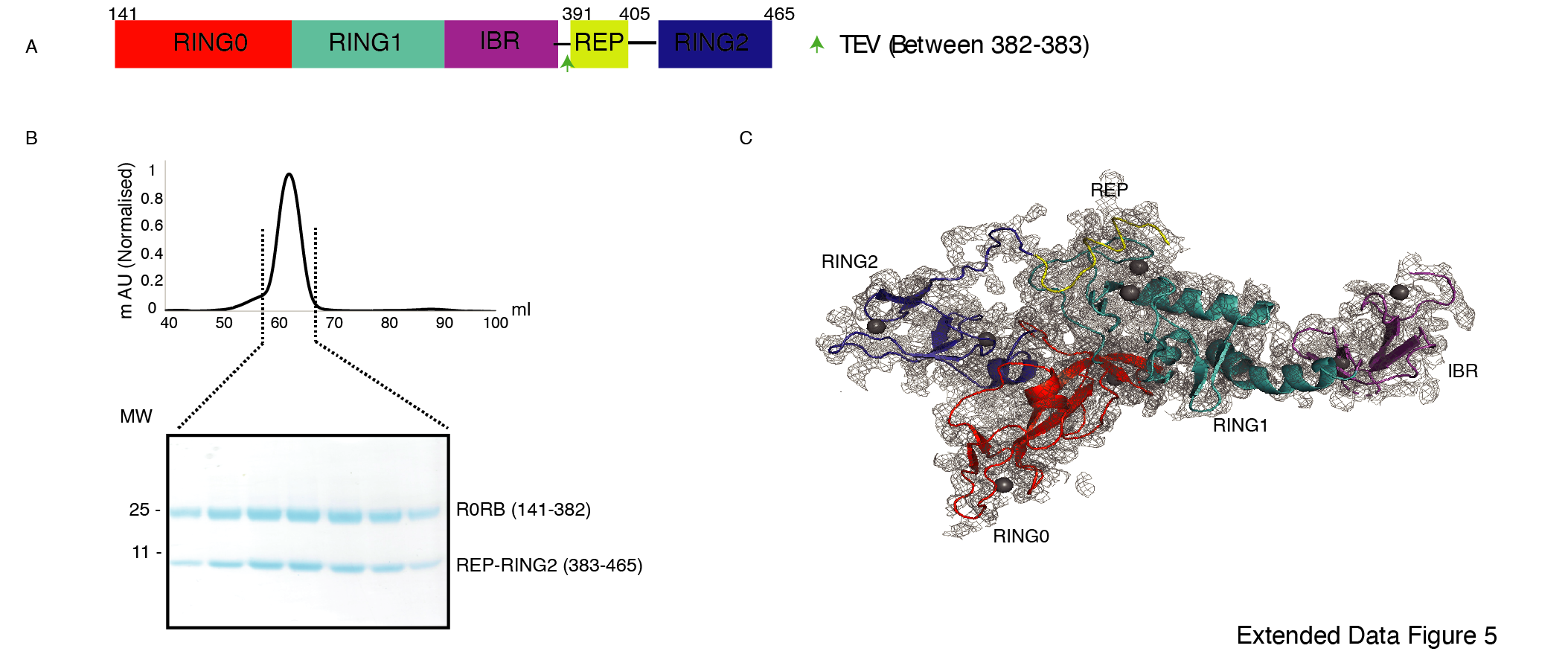
Further, to confirm that untethering does not affect the native interactions between RING2 and RING0 domains, we purified and determined the structure of TEV-treated R0RBR Parkin (141-465, TEV site between 382nd - 383rd, Extended Data Fig. 5A). Co-elution of R0RB (141-382) and RING2 (383-465) fragments on SEC (Extended Data Fig. 5B) and crystal structure showing intact native interactions between RING2 and RING0 (Extended Data Fig. 5C) further ruled out any possibility of artifact.
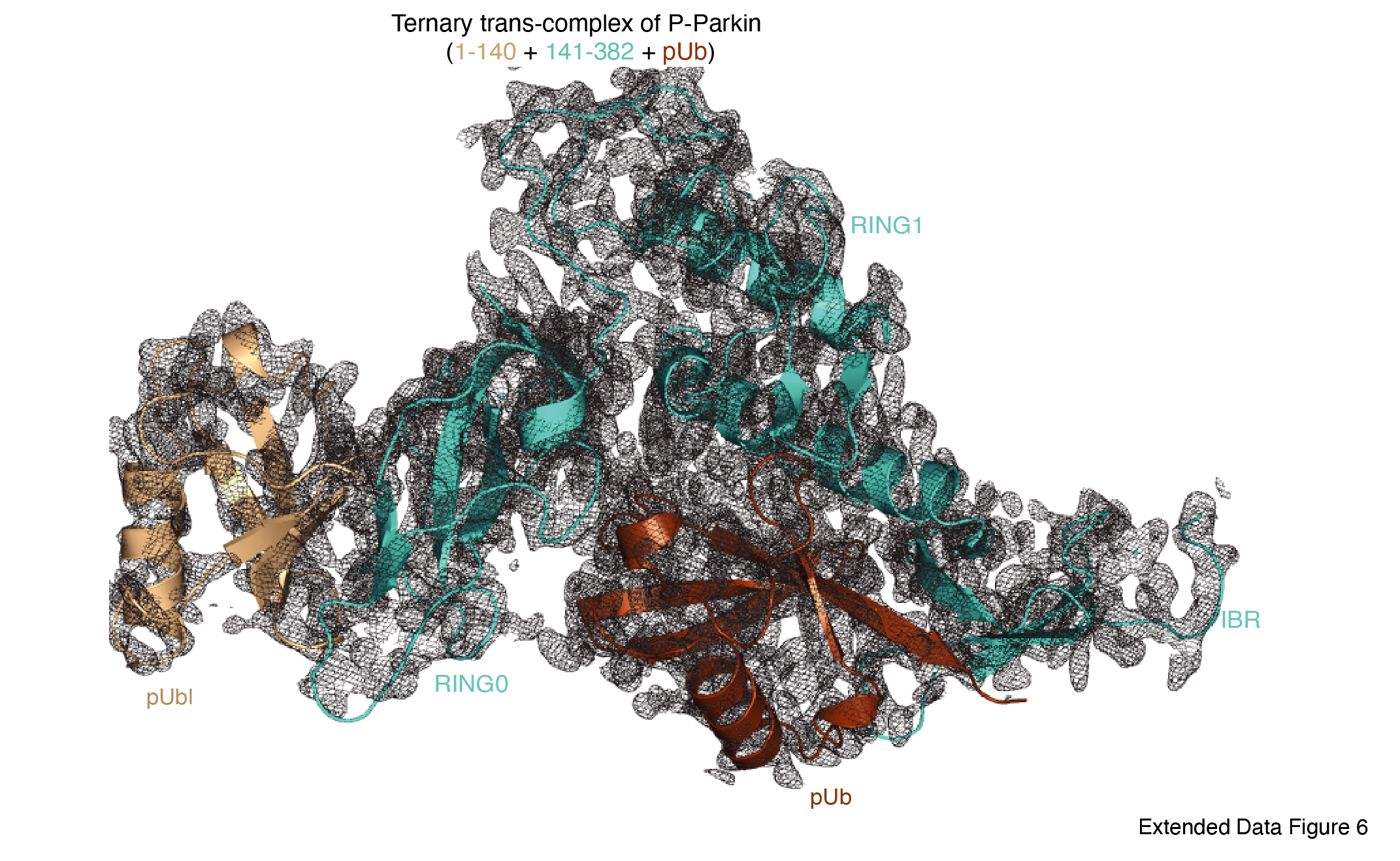
Additionally, we wanted to understand the molecular details of the complex seen in Figure 2B. We used FL-Parkin (Lys211Asn, HRV 3C site between 140th and 141st) as a donor of pUbl-linker (1-140), and R0RBR Parkin (TEV site between 382nd–383rd) as an acceptor of pUbl-linker. Phospho-Parkin (Lys211Asn) formed a stable complex with R0RBR protein with untethered RING2, and RING2 (383-465) was removed from R0RBR (Fig. 2C). The fractions containing the complex of phospho-Parkin (Lys211Asn) and R0RB (141-382) upon treatment with 3C followed by incubation with pUb showed co-elution of components of the ternary trans-complex (R0RB (141-382), pUbl-linker (1-140), and pUb) on SEC (Fig. 2C). The crystal structure of the ternary trans-complex of phospho-Parkin (pUbl-linker (1-140) + R0RB (141-382) + pUb) solved at 1.92 Å (Extended Data Table 1) further confirmed trans-complex formation between Parkin molecules (Fig. 2D, Extended Data Fig. 6). In the crystal structure, the pUbl domain from the donor molecule binds in the basic patch of RING0 of the acceptor molecule (Fig. 2D) in trans, similar to what is previously seen in the phospho-Parkin (1-382) structure with fused pUbl domain and untethered/truncated RING2 in cis molecule 24,29. Interestingly, the linker between pUbl and RING0 remained disordered in all the structures 24,29. Therefore, it would be difficult to say whether, in the previous cis structure, the pUbl bound to RING0 was from the same molecule or different molecules. Also, the fusion of pUbl with RING0 and untethering/truncation of RING2 as in the previous structures 24,29 may favor pUbl binding with RING0 in cis. Our data establish that keeping pUbl and RING2 untethered from their binding partner RING0, thus reducing the artifact due to the higher net concentration of the fused domain with RING0, is ideal for measuring trans interactions using biophysical methods.
Phospho-Parkin activates native Parkin in trans
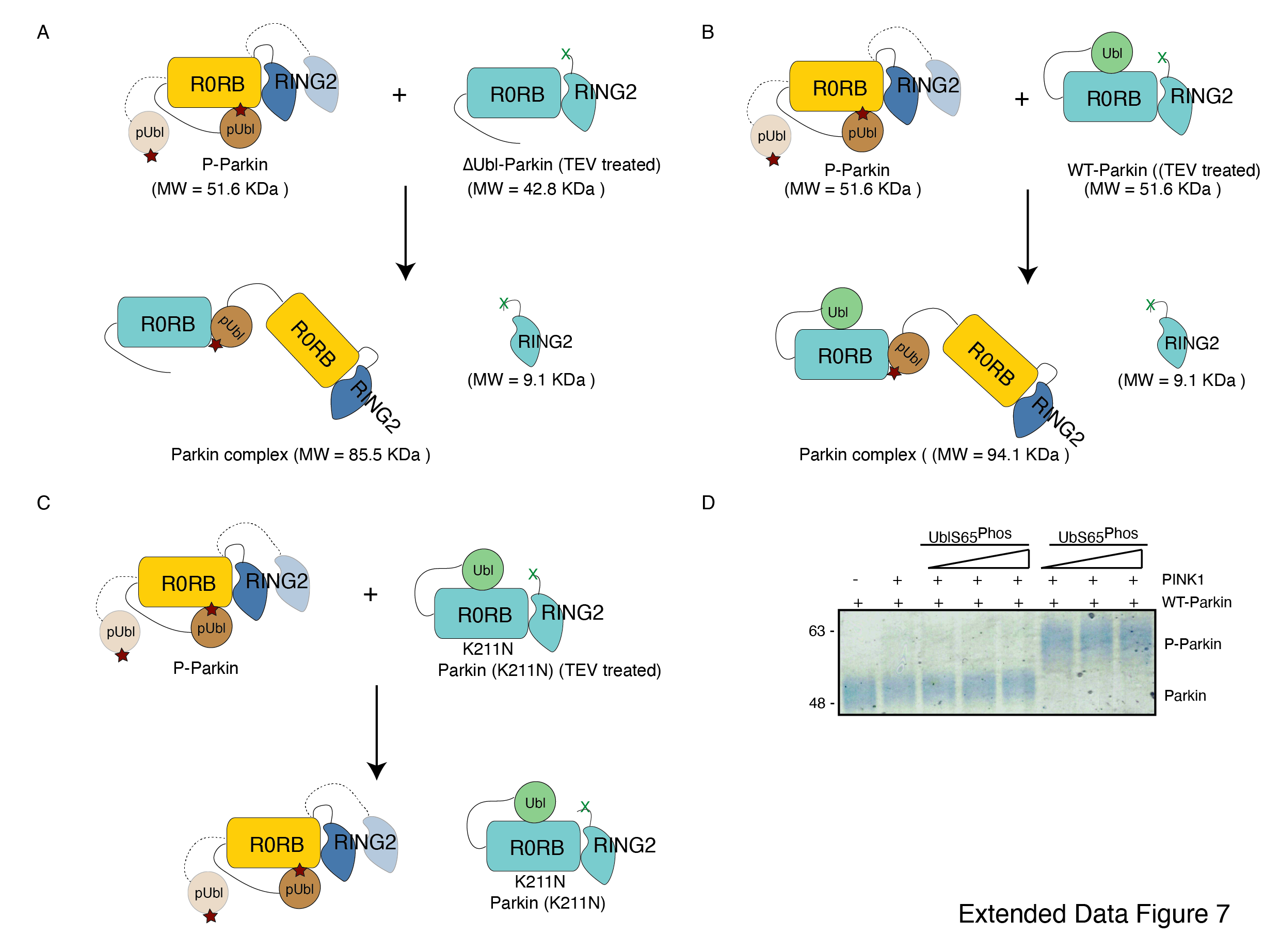
As the pUbl domain remains dynamic in both native phospho-Parkin and phospho-Parkin Lys211Asn (Extended Data Fig 2C, Extended Data Fig 4A), we wondered whether a trans-complex is formed using native phospho-Parkin. To test this, we used native phospho-Parkin (1-465) as a pUbl donor and ∆Ubl-Parkin (77-465, TEV treated) as a pUbl acceptor on RING0. Interestingly, phospho-Parkin forms a stable complex with ∆Ubl-Parkin (77-382) and results in the removal of RING2 (383-465) from ∆Ubl-Parkin (Fig. 3A, Extended Data Fig. 7A). We further tested the binding of phospho-Parkin (1-465) with WT-Parkin (1-465, TEV treated) as a pUbl acceptor. Similar to ∆Ubl-Parkin, phospho-Parkin forms a stable complex with WT-Parkin (1-382) and results in the removal of RING2 (383-465) from WT-Parkin (Fig. 3B, Extended Data Fig. 7B). However, mutation in the basic patch of Parkin (1-465, Lys211Asn, TEV treated) perturbed the complex formation with phospho-Parkin (Fig. 3B, Extended Data Fig. 7C) confirming that interactions between pUbl and the basic patch on the RING0 domain form trans-complex. We also confirmed complex formation using SEC-MALS (size-exclusion chromatography coupled with multi-angle light scattering) to rule out any ambiguity. MALS analysis further confirms complex (Observed M. W. = 94 ± 3 Kda) formation when phospho-Parkin (Observed M. W. = 53 ± 2 Kda) was mixed with TEV-treated WT-Parkin (Observed M. W. = 52 ± 3 Kda) (Fig 3C, Extended Data Fig. 7B).
As our binding experiments suggested that phosphorylated Parkin can bind with unphosphorylated Parkin, we wanted to check whether phosphorylated Parkin can activate unphosphorylated native Parkin. To test phospho-Parkin mediated native Parkin activation in trans, we used a catalytic-inactive version of phospho-Parkin with mutations in both the E2 binding site (T270R) and catalytic site (C431A). Interestingly, we observed that WT-Parkin ubiquitination/autoubiquitination activity is increased with increasing concentrations of phospho-Parkin (T270R, C431A) (Fig. 3D). Although, we were not expecting activation of WT-Parkin by phospho-Parkin as Ubl of WT-Parkin would block the E2 binding site on RING1 in WT-Parkin, activation of WT-Parkin with phospho-Parkin (T270R, C431A) suggested that a significant inhibition on Parkin is mediated by RING0 blocking RING2, which is released upon binding with pUbl. Further, we wondered whether pUbl could also enhance Parkin phosphorylation similar to what is seen previously with pUb 32. To test this, we checked Parkin phosphorylation by PINK1 in the presence of pUbl or pUb. However, unlike pUb, pUbl did not affect the Parkin phosphorylation by PINK1 (Extended Data Fig. 7D), further confirming that pUbl and pUb binding lead to unique conformational changes in Parkin. Overall, this data shows pUbl domain-mediated dimerization of Parkin molecules leading to Parkin activation in trans.
Assessment of Parkin activation in cells
It has previously been reported that pUb may interact with the RING0 domain of Parkin and that loss of this interaction underlies loss of Parkin recruitment to the mitochondria in cells expressing Lys211Asn Parkin 39. However, we recently showed that pUb does not bind in the RING0 pocket and specifically bind with the RING1 pocket 33 that contrasts with phospho-Ubl binding in the RING0 pocket and displacing RING2 in trans (Fig. 3). Biophysical assays also reveal that unlike the tight binding of pUb in the RING1, pUbl binding in the RING0 pocket is very transient in nature. Furthermore, Lys211Asn mutation in the RING0 pocket results in loss of Parkin activity by both loss of pUbl-mediated interactions (Fig 3) and by Asn211-driven conformational changes leading to loss of Parkin activity independent of pUb binding 33. This loss of Parkin activity would lead to a reduced amount of pUb, resulting in loss of Parkin recruitment to mitochondria. Therefore, we decided to test for any activity-independent Parkin recruitment to impaired mitochondria using a Parkin translocation assay in HeLa cells 10,14-16,31. Consistent with previous studies, 10,14-16,31 full-length wild-type but not catalytic-inactive Cys431Phe GFP-Parkin is recruited to mitochondria following carbonyl cyanide m-chlorophenyl hydrazone (CCCP) treatment (Extended Data Fig. 8A-B). Similarly, we did not observe recruitment of GFP-Parkin Cys431Phe RING1 (His302Ala) or RING0 (Lys211Asn) mutants when expressed alone (Extended Data Fig. 8A-B).
However, we observed that co-expression of mCherry-tagged-Parkin WT with GFP-Parkin Cys431Phe enables GFP-Parkin Cys431Phe recruitment to the mitochondria, similar to a previous study 31(Fig. 4A, D). Under these assay conditions, we strikingly observed that mutation of the pUb binding pocket in the RING1 completely abolished recruitment of the double mutant GFP-Parkin Cys431Phe/His302Ala to the mitochondria, when co-expressed with mCherry-tagged-Parkin WT (Fig. 4B, D). This excludes a significant role for the RING0 pocket in pUb binding in the context of full length parkin expressed in cells following mitochondrial damage (Fig. 4B, D). In line with this, mutation of the RING0 binding pocket produced a moderate defect in recruitment of the double mutant GFP-Parkin Cys431Phe, Lys211Asn to the mitochondria when co-expressed with mCherry-tagged-Parkin WT (Fig. 4C, D), suggesting that the transient interaction between pUbl and RING0 of Parkin in trans acts in concert with pUb binding to RING1 pocket for optimal Parkin recruitment to sites of mitochondrial damage (Fig. 4C, D). Under all transfection conditions, we did not observe a significant difference in mCherry-tagged Parkin WT (Extended Fig. 8C). Furthermore, co-expression of GFP-Parkin Cys431Phe or GFP-Parkin Cys431Phe/Lys211Asn or GFP-Parkin Cys431Phe/His302Ala with the non-phosphorylatable mCherry-tagged-Parkin Ser65Ala failed to rescue recruitment to the mitochondria (Fig. 4A, B, C, D). These findings are in line with our biophysical data and highlight the importance of phospho-Ubl domain-mediated interactions in Parkin recruitment to the mitochondria.
ACT improves Parkin enzyme kinetics
A previous study identified a small region (101-109) in the linker between Ubl and RING0 as an activator element (ACT) required for Parkin activity 24. To further explore the role of the ACT, we tested whether the omission of ACT affects the binding of Parkin with the charged state of E2 (E2~Ub). We observed that phospho-Parkin in a complex with pUb binds tightly with E2~Ub and co-elutes on SEC (Fig. 5A). Interestingly, deletion of the ACT did not affect the complex formation with E2~Ub, as phospho-Parkin (∆ACT) in complex with pUb co-elutes with E2~Ub on SEC (Fig. 5A). As the displacement of RING2 is a crucial process during Parkin activation, we tested whether the removal of the ACT affects the displacement of the RING2 domain using our TEV-based SEC assay. We observed that phospho-Parkin (∆101-109/ACT) after treatment with TEV results in a shift where RING2 (383-465) is displaced from the Parkin core (1-382, ∆ACT), resulting in the elution of two fragments of Parkin separately on SEC (Fig. 5B). As the deletion of ACT did not show any functional defect in Parkin, we hypothesized that the presence of ACT at the interface of RING0 and RING2 might affect the dynamic nature of RING2 thereby regulating the enzyme kinetics. To test this hypothesis, we compared the kinetics of phospho-Parkin (∆ACT) ubiquitination activity over different time points. We observed that the deletion of ACT slows the kinetics of Parkin activity, doubling the time for phospho-Parkin (∆ACT) to reach a similar level of activity as un-modified phospho-Parkin (Fig. 5C).
ACT is more efficient in cis than in trans
We next examined the interactions between ACT and Parkin in our ternary trans-complex of phospho-Parkin (1-140 + 141-382 + pUb) structure solved at a similar resolution and in the same space group as the previous solved structure of phospho-Parkin (1-382) in complex with pUb 24. However, we do not see any density of the ACT region in the ternary trans-complex of phospho-Parkin (1-140 + 141-382 + pUb) structure (Fig. 6A, Extended Data Fig. 9A), unlike the phospho-Parkin (1-382) in complex with pUb structure where the ACT region was clearly shown to occupy the hydrophobic pocket on RING0 (Fig. 6A). Interestingly, we observed that in the ternary trans-complex of phospho-Parkin structure, Lys48 of the pUbl domain occupies the same pocket that Arg104 of the ACT region occupied in the phospho-Parkin in complex with pUb structure (Fig. 6A, Extended Data Fig. 9A). Also, the side-chain of Lys48 of the pUbl domain was disordered in the previous structure of phospho-Parkin (1-382) in complex with pUb (Fig 6A).
We wondered whether the lack of density in the ACT region was due to the preference of ACT to remain associated with the cis molecule rather than to be complemented by the trans molecule. To test this hypothesis, we determined the crystal structure of the ternary trans-complex of phospho-Parkin with cis ACT using phospho-Ubl (1-76) and ∆Ubl-Parkin (77-465, TEV site between 382nd - 383rd). pUbl formed a stable complex with ∆Ubl-Parkin (77-382) after TEV treatment and resulted in the displacement of RING2 (383-465) (Fig. 6B). Fractions containing trans-complex of phospho-Parkin with cis ACT (1-76 + 77-382) were mixed with pUb to get the crystals of the ternary complex. The ternary trans-complex of phospho-Parkin with cis ACT was crystallized, and structure was determined at 2.6 Å (Extended Data Table 1). Interestingly, in the structure of the ternary trans-complex of phospho-Parkin with cis ACT, we could observe the electron density in the ACT region (Fig. 6C, Extended Data Fig. 9B). Furthermore, Lys48, which occupied the ACT region in the ternary trans-complex of phospho-Parkin structure, was disordered in the ternary trans-complex of phospho-Parkin with cis ACT structure, similar to what is seen in the phospho-Parkin structure (Fig. 6A, C, Extended Data Fig. 9B).
To validate crystal structures, we compared the activity of R0RBR (141-465) and ∆Ubl-Parkin (77-465) with/without pUb, which showed that the presence of the linker containing ACT in ∆Ubl-Parkin (77-465) makes it more active compared to R0RBR (141-465) in both ubiquitination/auto-ubiquitination and Miro1 ubiquitination assays (Extended Data Fig. 9C). We then compared the activation of R0RBR and ∆Ubl-Parkin using pUbl (1-76) in trans. We observed that pUbl (1-76) efficiently activated ∆Ubl-Parkin (77-465); however, R0RBR (141-465) activation by pUbl (1-76) was very poor (Fig. 6D, Extended Data Fig. 9D). Further, we tested whether pUbl-linker (1-140) with or without ACT would affect the activation of ∆Ubl-Parkin (77-465) in trans. Interestingly, ubiquitination assays performed using increasing concentrations of pUbl (1-76) or pUbl-linker (1-140), or pUbl-linker-∆ACT (1-140, ∆101-109) showed that ∆Ubl-Parkin activation is not affected by the linker (77-140) or ACT region in trans (Fig. 6E). However, compared to pUbl (1-76), pUbl-linker (1-140) showed better activation of R0RBR (141-465) (Fig. 6F, Extended Data Fig. 9E). Also, in contrast to pUbl-linker (1-140), pUbl-linker-∆ACT (1-140, ∆101-109) showed poor activation of R0RBR (141-465) similar to pUbl (1-76) (Fig. 6F, Extended Data Fig. 9E). However, the activity of R0RBR (141-465) complemented with pUbl-linker (1-140) was less than the activity of ∆Ubl-Parkin (77-465) complemented with pUbl (1-76) (Fig. 6F, Extended Data Fig. 9E). Overall, our data suggest that ACT can be complemented in trans; however, it is more efficient in cis.
The crystal structure of pUbl-linker (1-140) depleted Parkin (141-465, Arg163Asp, Lys211Asn) in complex with phospho-ubiquitin reveals a new ubiquitin-binding site on Parkin
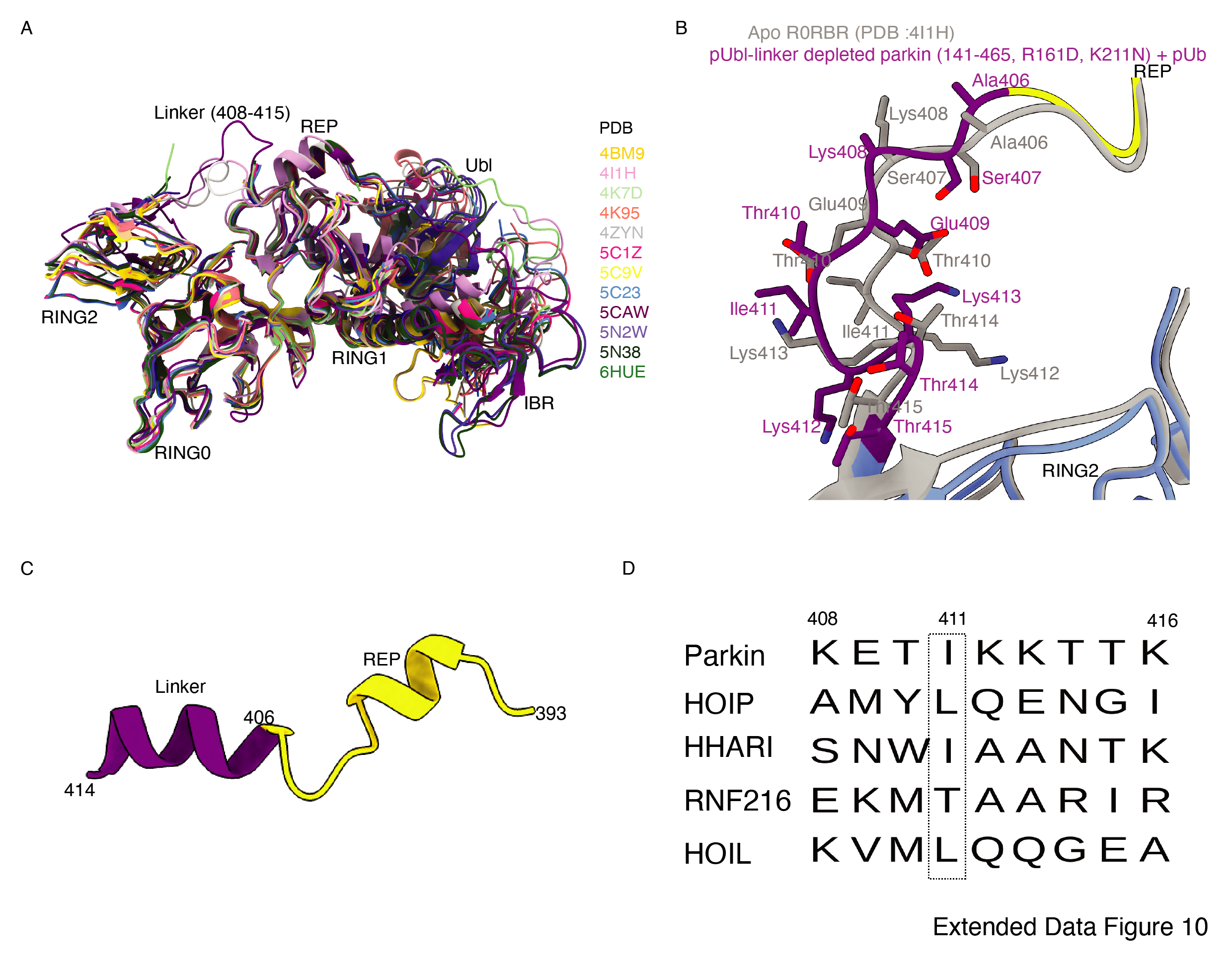
In the last few years, several structures of Parkin or Parkin complexes were solved in various conditions and from different species. However, the linker (408-415) between REP element and RING2 is mostly disordered, except in structures (PDB 4I1H, 5CAW, 4ZYN) where the above region is modelled in different conformations (Extended Data Fig. 10A), highlighting its flexible nature. A pathogenic mutation Thr415Asn is also found in the linker (408-415), which abolishes Parkin activity. However, the role of this small linker region on Parkin remains elusive. Therefore, we decided to closely inspect all the structures solved in the present study. We noticed that in pUbl-linker (1-140) depleted Parkin (141-465, Arg163Asp, Lys211Asn) in complex with pUb structure, out of two molecules of Parkin in the asymmetric unit, one molecule of Parkin shows good electron density in the linker (408-415) region of Parkin (Fig. 7A, B). We further noticed that in the structure of the pUbl-linker (1-140) depleted Parkin (141-465, Arg163Asp, Lys211Asn) in complex with pUb, the linker (408-415) undergoes conformational changes compared to the previously solved apo R0RBR structure (PDB 4I1H) (Extended Data Fig. 10B). Thr410, Ile411 and Lys412 facing outwards in the apo R0RBR structure, are present in the core of pUbl-linker (1-140) depleted Parkin (141-465, Arg163Asp, Lys211Asn) in complex with pUb structure (Fig. 7B, Extended Data Fig. 10B). Interestingly, we noticed interactions between the linker (408-415) of Parkin and phospho-ubiquitin from the neighboring molecule in the asymmetric unit (Fig. 7C). The core of interactions between the Parkin linker and ubiquitin is mediated by Ile411, which is involved in hydrophobic interactions with the hydrophobic pocket of ubiquitin (Fig. 7C). Other interactions between Parkin and ubiquitin include ionic interactions mediated by Lys412, and His422 (Fig. 7C). Water-mediated interactions between linker (408-415) and ubiquitin are also seen wherein Thr410 interacts with the carbonyl group of Arg72 of ubiquitin, and Thr415 interacts with the carbonyl of Gly35 of ubiquitin (Fig. 7C). Furthermore, Glu409 forms a salt bridge with Lys413 (Fig. 7C), which could be required for maintaining the structure of the linker region for ubiquitin binding. Also, residues in the linker interacting with ubiquitin are highly conserved in Parkin across different species (Fig. 7D), suggesting their functional importance. Our data in Fig. 1 suggests that RING2 remains flexible in the open state after pUbl binding in the basic patch. As the current structure is captured in the closed state, we wondered whether the linker connecting REP and RING2 may adopt an alternate conformation. The crystallization of the open state of phospho-Parkin remains challenging due to the flexible/multiple possible conformations of the REP-RING2 region. Therefore, we used AlphaFold 34 to predict the model of the linker region of Parkin. Interestingly, the AlphaFold model shows helical structure in the linker region of Parkin (Extended Data Fig. 10C), further confirming the flexible nature of this region.
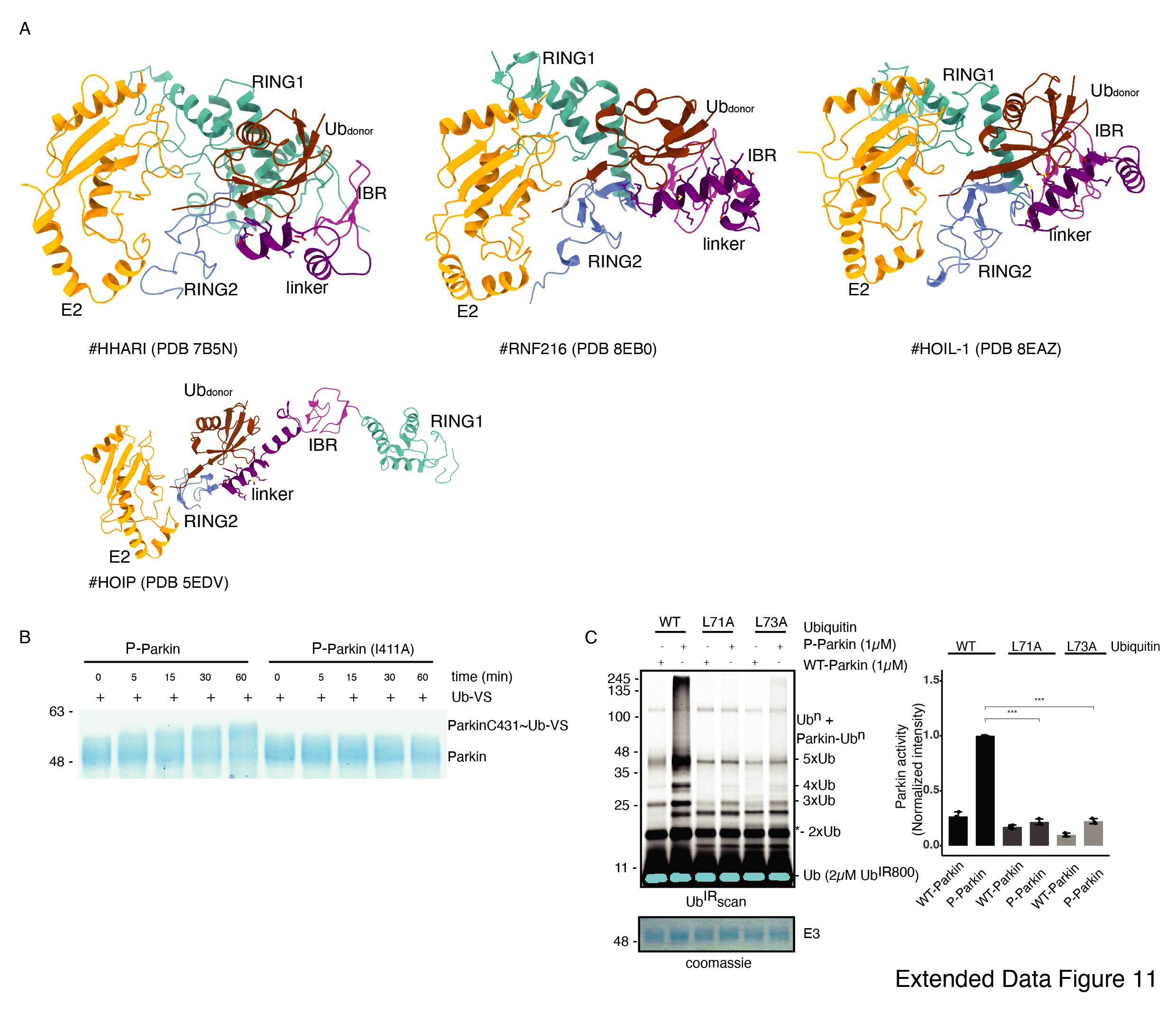
To validate the observations from structural analysis, we mutated these residues and compared their ubiquitination activity. In contrast to WT-Parkin, mutation of Lys409 and His422 drastically reduced Parkin activity, whilst Ile411Ala, Thr415Asn, and Lys416Ala resulted in the complete abolishment of Parkin activity (Fig. 7E). Further inspection revealed that although the linker region of Parkin is not conserved across different members of RBR family E3-ligases (Extended Data Fig. 10D), hydrophobic nature at the corresponding position of Ile411 on Parkin is conserved among various RBRs except RNF216 (Extended Data Fig. 10D). The crystal structures of HOIP, HOIL, HHARI, and RNF216 solved with E2~Ub 35–37 show linker region interacts with donor ubiquitin (Ubdon) (Extended Data Fig. 11A). To test whether the linker between REP and RING2 of Parkin binds with donor ubiquitin (Ubdon), we performed binding assays using E2~Ub. Interestingly, unlike phospho-Parkin, which forms a stable complex with E2~Ub on SEC and co-elutes with E2~Ub and phospho-ubiquitin (Fig. 7F), Ile411Ala mutation completely abolished Parkin interaction with E2~Ub (Fig. 7F). Furthermore, phospho-Parkin Ile411Ala was unable to be charged by Ub-VS (Extended Data Fig. 11B). We also tested Parkin activity using ubiquitin mutants (Leu71Ala or Leu73Ala) which would perturb the interactions of ubiquitin and Parkin linker as suggested by structure in Fig. 7C. Compared to native ubiquitin, ubiquitin mutants show loss of Parkin activity (Extended Data Fig. 11C) which nicely corroborates with our data. Overall, our data show that the linker region between REP and RING2 interacts with donor ubiquitin and plays a crucial role in Parkin function.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}