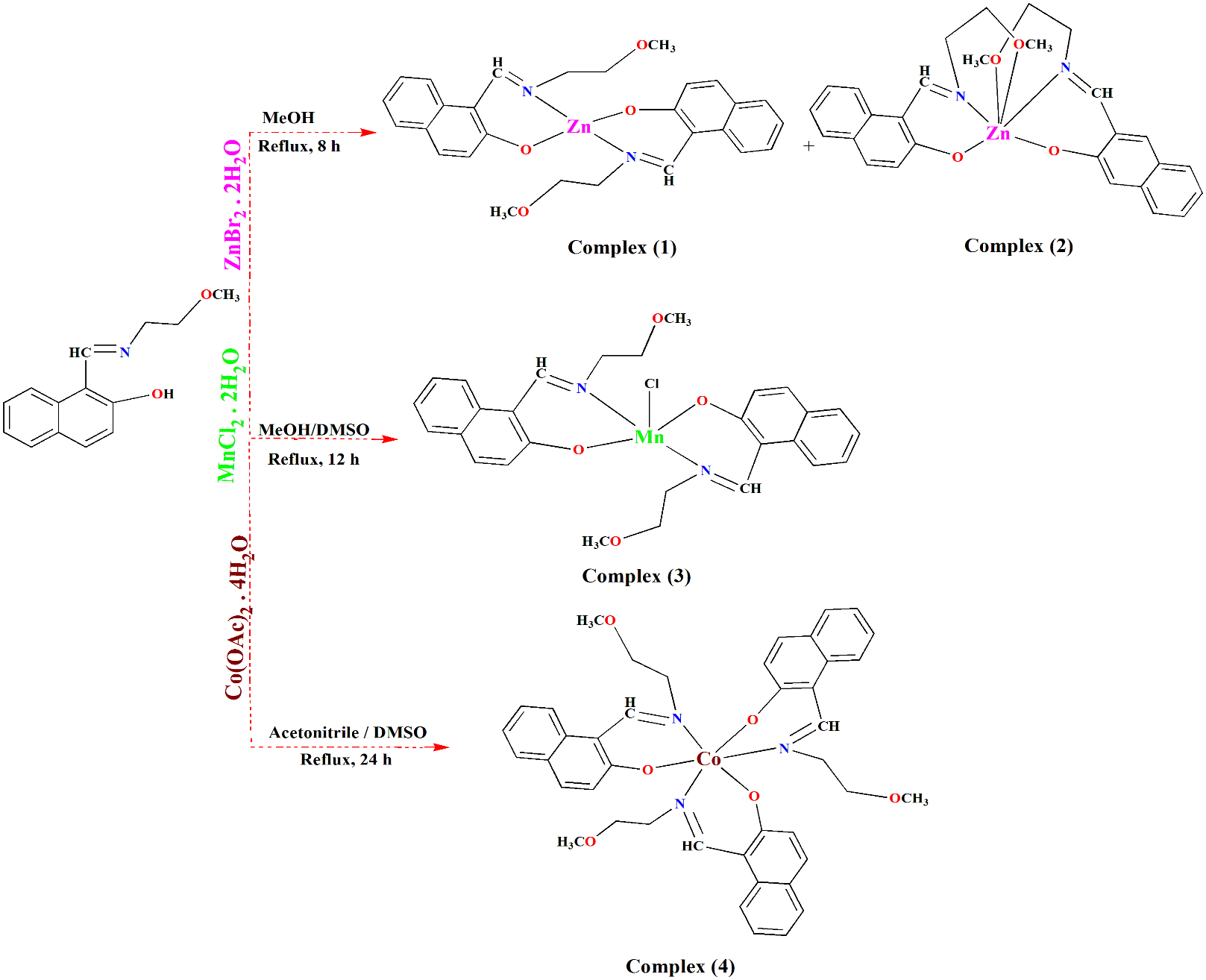
3.1. Synthesis
Schiff base complexes were prepared from the reaction of ligand HL with methanolic solution containing metal salt with a ratio of 1:1 ZnBr2.2H2O (1), MnCl2.2H2O (2), and Co(OAc)2.4H2O (3). The complexes were identified using spectroscopic and crystallographic techniques. Also, they showed good elemental analysis. The electrochemical properties of the complexes indicate the electronic effects of the groups and the redox potential. Hirshfeld surface analysis was done to study and understand short contacts and non-covalent interactions, and with the help of graphic displays based on Hirshfeld surface analysis, di vs de mapped were also calculated. Furthermore, in this study, is discussed computational chemistry and molecular docking studies.
3.2. FT-IR spectra
In the FT-IR spectrum of the ligand, a strong peak in the region of 1627 cm− 1 indicates the formation of the imine group (C = N). The observed peak in the 1438 region was attributed to the stretching vibration of the (C─N) group (Figure S1). According to the FT-IR spectrum of the Schiff base complexes in Figure S2, S3 and S4, it has a shift compared to the stretching vibration bands in the ligand, which confirms the synthesis of the complex. The stretching vibration band of the C = N group in these complexes was observed at a wave number lower than that of the ligand in the regions of 1618 (1, 2), 1616 (3), and 1606 (4) cm− 1[14, 15]. The stretching vibration band in the regions of 1355 (1, 2), 1361 (3) and 1361 cm− 1 (4) is attributed to the C─O band. Also, the observed shift of the complex (1–4) compared to the ligand is determined at 1461 (1, 2), 1454 (3) and 1436 cm− 1 (4) corresponding to the stretching vibration of the C─N group. In addition,
metal-nitrogen and metal-oxygen absorption bands appeared in the regions of 421–580 cm− 1 (1, 2), 481–572 cm− 1 (3) and 414–524 cm− 1 (4), respectively. The broad absorption band in the regions of 2760–3190 cm− 1 was attributed to C─H stretching vibration [14, 15, 31–33].
Figures. S1-4.
3.3. UV–Vis spectra
In the electronic spectrum of the ligand, the three peaks at the wavelength of 306, 400 and 420 nm indicate the transitions (C = N) π → π* and n → π*(Fig.S5). The electronic spectrum of the collected complexes recorded in the acetonitrile solution and is shown in the Fig.S6-8. The absorption band observed in the complex (1–4) in the area 231 and 250 nm (1, 2), 230 and 249 nm (3), 208 and 231 nm (4) was attributed to the aromatic ring π → π*. Also, the absorption band appeared at 304 and 344 nm (1, 2), 306 and 330 nm (3), 275 and 322 nm (4) related to the transmission of intra -ligand (C = N), which has shifts compared to the ligand. The bands appeared in the area 403 and 418 nm (1, 2), 396 and 429 nm (3), 438 nm (4) are related to the charge transfer. For complexes (3, 4) in the 484 and 675 nm districts, d → d transfers was observed, respectively [13–15, 34–36].
Figures. S5-8.
3.4. Description of the crystal structures
Perspective views of the complexes are shown in Figs. 1–4; most important geometrical data are listed in Table 2. Interestingly, complexes display different coordination modes. Complex 1 of identical ZnL2 composition is different, as it is unsymmetrical and the central ion is coordinated by three atoms from each L molecule (N2O3). It has to be noted, however that two additional Zn…O contacts are much longer than the other bonds. So, the coordination number in this case is six and the environment is slightly deformed octahedron. In this structure additionally, a solvent – methanol molecule was found, connected by hydrogen bond with O1A atom (O···O 2.721 Å, H···O 1.89 Å, O-H···O 172º). Complex 2, ZnL2, is C2-symmetrical (Zn ion lies on the twofold axis), and the central atom is coordinated by two oxygen and two nitrogen atoms (N2O2) from the ligand molecules in quite regular tetrahedral fashion.
Compound 3, Mn complex, has the MnL2Cl structure and is again C2-symmetrical (Mn and Cl atoms lie on the twofold axis). Mn is coordinated by two nitrogen atoms and two oxygen atoms from both L molecules and additionally by Cl, which occupies an apex of the tetragonal pyramid (c. n. 5). Finally, Co complex 4 has the composition CoL3, one nitrogen and one oxygen atom from each ligand is involved in coordination which gives the coordination number 6 and octahedral geometry.
It might be noted that both the geometries of the ligand molecules and coordination schemes are quite typical for similar complexes. In the crystallographic database CSD there are 1727 structures of Zn complexes with c.n 4 and N2O2 coordination, 549 for Zn with c.n. 6 and N2O3, 92 for Mn with c.n. 5 and N2O2Cl and 1217 of Co, c.n. 6, N3O3.
Table 2
Relevant geometrical parameters (Å, º) with s.u.’s in parentheses
| |
1 (M = Zn)
|
2 (M = Zn)
|
3 (M = Mn)
|
4 (M = Co)
|
|
M1-O1A
|
1.992(4)
|
1.921(7)
|
1.852(2)
|
1.8837(15)
|
|
M1-N12A
|
1.993(5)
|
1.969(8)
|
2.045(3)
|
1.9341(17)
|
|
M1-O15A
|
2.451(4)
|
|
|
|
|
M1-O1B
|
1.961(4)
|
1.921(7)
|
1.852(2)
|
1.8905(14)
|
|
M1-N12B
|
1.992(5)
|
1.969(8)
|
2.045(3)
|
1.9359(17)
|
|
M1-O15B
|
2.546(4)
|
|
|
|
|
M1-O1C
|
|
|
|
1.9008(15)
|
|
M1-N12C
|
|
|
|
1.9395(17)
|
|
M1-Cl1
|
|
|
2.3890(16)
|
|
|
O1A-M1-N12A
|
90.50(18)
|
94.4(3)
|
87.38(10)
|
|
|
O1A-M1-O1B
|
105.61(18)
|
110.2(4)
|
170.19(17)
|
173.90(6)
|
|
O1A-M1-N12B
|
101.45(18)
|
120.9(3)
|
89.69(11)
|
|
|
N12A-M1-O1B
|
104.68(17)
|
120.9(3)
|
89.69(11)
|
|
|
N12A-M1-N12B
|
156.4(2)
|
117.9(5)
|
145.28(16)
|
|
|
O1B-Mn1-N12B
|
91.69(18)
|
94.4(3)
|
87.38(10)
|
|
|
O1A-M1-O15A
|
162.85(16)
|
|
|
|
|
O1A-M1-O15B
|
84.72(16)
|
|
|
|
|
N12A-M1-O15A
|
75.12(17)
|
|
|
|
|
N12A-M1-O15B
|
87.83(17)
|
|
|
|
|
O1B-M1-O15A
|
87.36(17)
|
|
|
|
|
O1B-M1-O15B
|
163.48(16)
|
|
|
|
|
N12B-M1-O15A
|
89.09(17)
|
|
|
|
|
N12B-M1-O15B
|
73.44(17)
|
|
|
|
|
O1A-M1-Cl1
|
|
|
94.91(8)
|
|
|
N12A-M1-Cl1
|
|
|
107.36(8)
|
|
|
O1B-M1-Cl1
|
|
|
94.91(8)
|
|
|
N12B-M1-Cl1
|
|
|
107.36(8)
|
|
|
O1C-Co1-N12B
|
|
|
|
175.58(7)
|
|
N12A-Co1-N12C
|
|
|
|
175.95(7)
|
|
C10-C11-N12-C13
|
174.9(5)
176.9(6)
|
176.7(9)
|
-171.0(3)
|
176.4(2)
178.9(2)
-168.6(2)
|
|
C11-N12-C13-C14
|
134.7(5)
130.3(6)
|
-104.6(11)
|
87.5(4)
|
-81.9(2)
92.6(2)
80.7(2)
|
|
N12-C13-C14-O15
|
54.1(6)
56.7(7)
|
65.0(13)
|
-66.1(4)
|
62.9(2)
-60.2(3)
-69.5(3)
|
|
C13-C14-O15-C16
|
176.5(5)
-178.2(6)
|
-172.3(12)
|
165.6(3)
|
171.75(19)
179.4(2)
-78.1(3)
|
Figure. 1–4
Table 2
3.5. Electrochemical studies
Analysis of the electrochemical behavior of the synthesized complexes was recorded using cyclic voltammetry in DMSO solution. As shown in Figs. (5–7), the synthesized complexes (1–4) have shown irreversible behavior. In the cyclic voltammogram of the complex (1 and 2), a reduction process in the cathodic potential (Epc) was observed in the area of -1.16 and 0.219 V, and these areas are assigned to the reduction of ZnII/I and ZnI/0. According to Fig. 6 for complex (3), the cathodic voltagram observed at the potential of -0.84 and − 1.52 V indicates MnIII/MnIІ and MnII/MnІ. Cyclic voltammetry in Fig. 7 shows that in the cyclic voltammogram for complex (4), a decreasing voltammogram in cathodic potential (Epc) -0.531 and − 1.34 V caused by CoШ/CoII and CoII/CoI has been observed. In addition, the redox process observed at the potential of 0.36 V is related to CoII/CoIII oxidation [15, 37–39].
Figure. 5–7
3.6. Investigation of intermolecular interactions by HS analysis and FPs plot
The HSA facilitates visualization of the molecular component by permitting surface transparency via the use of dnorm, shape index, and fingerprint plots. The color code on Hirshfeld surfaces represents the geometrical function dnorm for complexes (1–4). The red patches on the dnorm surface denoted interatomic or shorter connections (< vdW radii). The graphics clearly show that the darker the color of the red region, the stronger the interactions, and the other shallower areas represent mostly the distribution of weaker the exchanges. The distribution of the approximate hydrogen bonding in the complexes may be evaluated using these figures, which is useful in further exploring the intrinsic aspects of the complex's stable existence.
3.6.1. HSA and FPs of the complex [ZnII(HL)2 CH3OH] (1), [ZnII(HL)2] (2)
For complex [ZnII(HL)2CH3OH] (1), the red spots obvious with dnorm revealed dominant interactions on the surface of O···O, C···H/H···C and O···H/H···O that contribute significantly to complex (1). Figure 8a, b labelled 1–14 observable red spots of 1 and 2 on account of two pairs of intermolecular C3A–H3A···O1C(number:1) and C5A–H5A···O1C(number:2) hydrogen bonds (labeled as a) whereas intermolecular H1C··· O1C (number:11) hydrogen bond (labeled as b), also, intermolecular C13D–H13D···C7B (No:3), C13D–H13D···C8B(No:4), C14A–H14A···C4B (No:9), C14A–H14A···C9B (No:10), C13C–H13C···C9A (No:13) and C14C–H14C···C7A(No:14) short contacts were indicated by light red spot (labeled as a, b) between one (C–H)amine molecule and C atom of aromatic ring aldehyde. As well as, the C3B–H3B···C9A (No:6), C3B–H3B···C10A (No:7) and C8A–H8A···C5B (No:8) interactions via the (C–H)2−hydroxy 1−naphtaldehyde group with C atoms of the 2-hydroxy 1-naphtaldehyde groups are apparent as the light zone on the Hirshfeld surface. The dark red spots on the Hirshfeld surface suggest predominant interatomic and shorter contacts (C1A…H1C, No:5). Moreover, the O1C…O1A (No:12) contact is involved between the (O)2−hydroxy 1−naphtaldehyde atom of the complex and (O)MeOH solvent. For complex [ZnII(HL)2] (2), the exchanges stronger, are identified with red on the Hirshfeld surface (Fig. 9a, b), which are recognizable to an interaction with C…C, C―H…C and C―H…O hydrogen bonds. The light red spots on the Hirshfeld surface shown C2…C16 (No:1), C8…C3 (No:2) (labeled as a) contacts between two the (C)2−hydroxy 1−naphtaldehyde group with (C)amine group (for No:1) and via two the (C)2−hydroxy 1−naphtaldehyde group (for No:2). Furthermore, the light red spots on the surface indicated C6―H6…O15 (No:4) and H3―C3…C16B (No:5) interactions (labeled as a, b). In addition to the discussed short contacts, one of the dark red spots on the Hirshfeld surface indicates C13―H13A…O1 (No:3), (labeled as a, b) hydrogen bond between (O)2−hydroxy 1−naphtaldehyde group and (C―H)amine molecule.
Figures. 8–9
The shape index schemed on the Hirshfeld surface for complexes [ZnII(HL)2CH3OH] (1) and [ZnII(HL)2] (2) molecules in Figs. 10(a, b) indicates that the aromatic ring (C…C) interactions between neighboring molecules labeled with number 1 on the shape index are blue- red bow-tie designs. Also seen in this figure, the “red π -holes”, which represent C—H··· π interactions, are labeled 2. These interactions are caused by C—H contacts with the center of the aromatic ring [22, 40].
Figures. 10
In order to better understand the contacts in the structure of the studied complexes, the fingerprint diagrams of FPs were examined (Figs. 11 and 12). In both complexes (1) and (2), it was shown that H...H contacts cover the most part of the fingerprint diagrams (54.5% and 53.6%) and with light colored parts from blue to blue-green, about de=di 1.1 to 1.8 Å for (1 and 2) it includes.
In addition to this contact, a pair of wings was observed in the FPs graphs, which was created by the combination of C...H/H...C contacts, and it made up about 35.7% and 32.3% for (1 and 2) of the surface of this graph. These combined contacts lead to the formation of C—H... π interactions (the shortest contacts were obtained with minimum values of di + de ≈2.6 and 2.8 Å). In addition, the two prongs observed in the diagram predict C—H⋯O hydrogen interactions in the structure of both complexes 1 and 2, which are formed by O⋯H contacts (number 3). This interaction occupies about 8.9% (for 1) and 11.3% (for 2) of the fingerprint diagram. C—H...O interactions have shown contact with minimum values of di + de < 1.2–2.6 Å (asymmetrically for 1) and 2.0 Å (symmetrically for 2). In the structure of these complexes, weaker contacts with a percentage of less than 2 are observed, contacts such as C...N(0.2%), O...N(0.2%), C...C (0.2%) and N...H (0.1%) cover the surface of the chart.
Figure. 11–12
3.6.2. HSA and FPs of the complex [(MnIII(HL)2) Cl](3)
The dark and light red areas appeared by dnorm function confirm several types of interactions and non-classical hydrogen interactions such as: C…C, C―H…C and C―H …Cl (Fig. 13). The C2...C17 (No:1) interaction between two neighboring carbon groups of the 2-hydroxy 1-naphthaldehyde ring and 2-ethoxyethylamine was seen as a bright red area on the Hirshfeld surface. In addition, the C4―H4…C11 (No:2) contact between the carbon molecule of the (C)HC=N group and the carbon molecule of (C) 2−hydroxy 1−naphthaldehyde ring is revealed in the bright area. Furthermore, the dark red spot represents the C6-H6…Cl (No:3) hydrogen bond, which are formed by the interaction between the complex molecule with the (C)2− hydroxy 1−naphthaldehyde atoms and with the (Cl) atom (Fig. 13).
Figure. 13
Similar to the [ZnII(HL)2CH3OH] (1), [ZnII(HL)2] (2) complexes, the shape index confirms the presence of π…π (blue-red triangles) and C–H…π (red π-holes) interactions [22, 40], and is shown in Fig. 14.
Figure. 14
According to the calculation of the 2D diagrams in this complex, it can be understood that 93.1% of the total contact is made up of hydrogen atoms, which confirms the more important role of hydrogen atoms compared to others. Figure 15 shows the fingerprint diagram of complex (3), five types of H... interactions were calculated with different proportions: H...H (46.6% > H...C/H…C (33.6%) > H... O/O…H (8.9%) > H... Cl/Cl…H (1.9%) > H... N/N…H(1.8%). In this complex, similarly to [ZnII(HL)2CH3OH] (1), [ZnII(HL)2] (2) complexes, the number 1 in the graphs shows H...H type contacts, and short contacts were calculated with values of di + de ≈ 2.2 Å. C...H/H...C (number 2) contacts are shown as wings in the graph. C—H... π interactions that formed the shortest contacts with minimum values of di + de ≈ 2.8 Å. Number 3, O...H/H…O contacts that shows C―H...O hydrogen bonds show (di + de ≈ 2.8 Å). Other weak intermolecular exchanges include less than 1.8% of C…C (0.9%) and Mn…H/H…Mn (0.3%) is formed.
Figure. 15
3.6.3. HSA and FPs of the complex [CoIII(HL)3] (4)
By examining Fig. 16(a-c), the visible red spots on the Hirshfeld surface represent hydrogen bonds C—H…C, C—H…O, C…C, C…H and H…H in the [CoIII(HL)3] complex (4). The bright red spots correspond to C7A—H7A…C8B (No:3), C7A—H7A…C9B (No:4) and C16C—H16C…C4B (No:8) interactions, these interactions are due to contact and proximity between (C—H) 2−hydroxy 1−naphthaldehyde with (C) 2−hydroxy 1−naphthaldehyde atom (for No:3 and 4) and via (C—H) 2−ethoxyethylamine with (C) 2−hydroxy 1−naphthaldehyde atom (for No:8), were observed. Also, the hydrogen interactions with bright red dots represent C14D—H14D…O15A (No:5), C2C—H2C…O15C (No:9) and C3C—H3C…O15C (No:10), which are caused by the contact of the (O) 2−ethoxyethylamine atom with (C-H)2−hydroxy 1−naphthaldehyde (for No:9 and 10) and (C—H)2−ethoxyethylamine(for No:5). In addition, the contacts H5A…H16C (No:1), C2A... C4C (No:2), H10B…H16F (No:6) and C2C…H16A (No:7) were observed in Figs. 16a, b. Stronger interactions with dark red dots include (No:6) formed through short proximity of (H)2−hydroxy 1−naphthaldehyde and (H)2−ethoxyethylamine. Weaker interactions with bright red dots on surface also confirm the short contacts between the carbon atom of 2-hydroxy-1-naphthaldehyde and (C)2−ethoxyethylamine (for No:2), as well as, the carbon atom of 2-hydroxy-1-naphthaldehyde with (H)2−ethoxyethylamine (No:7). More information about the interactions between dark and light red spots for all complexes is summarized in Table 3.
Table 3
Close contacts to be prominent in the complexes.
|
D―H...A
|
d(D―H) (Å)
|
d(H...A) (Å)
|
d(D...A) (Å)
|
∠ (DHA) (°)
|
Label
|
Color code
|
|
[ZnII(HL)2CH3OH] (1)
|
|
C3A―H3A…O1C
|
0.949
|
2.575
|
3.441
|
151.76
|
(1)
|
light red spot
|
|
C5A―H5A…O1C
|
0.951
|
2.609
|
3.466
|
150.15
|
(2)
|
light red spot
|
|
C13D―H13D…C7B
|
0.950
|
2.733
|
2.650
|
94.96
|
(3)
|
light red spot
|
|
C13D―H13D…C8B
|
0.949
|
2.801
|
2.770
|
98.38
|
(4)
|
light red spot
|
|
C1A...H1C
|
-
|
2.787
|
-
|
-
|
(5)
|
dark red spot
|
|
C3B―H3B…C9A
|
0.951
|
2.728
|
3.604
|
153.50
|
(6)
|
light red spot
|
|
C3B―H3B…C10A
|
0.951
|
2.749
|
3.490
|
135.30
|
(7)
|
light red spot
|
|
H5B―C5B…H8A
|
0.950
|
2.740
|
2.829
|
96.50
|
(8)
|
light red spot
|
|
C14A―H14A…C4B
|
0.991
|
2.801
|
3.477
|
125.97
|
(9)
|
light red spot
|
|
C14A―H14A…C9B
|
0.991
|
2.826
|
3.731
|
152.21
|
(10)
|
light red spot
|
|
O1C...H1C
|
-
|
1.887
|
-
|
-
|
(11)
|
light red spot
|
|
O1A...O1C
|
-
|
-
|
2.721
|
-
|
(12)
|
light red spot
|
|
C14C―H14C…C7A
|
0.950
|
2.795
|
3.139
|
102.39
|
(13)
|
light red spot
|
|
C13C―H13C…C9A
|
0.990
|
2.764
|
3.588
|
140.97
|
(14)
|
light red spot
|
|
[ZnII(HL)2] (2)
|
|
C2…C16
|
-
|
-
|
3.329
|
-
|
(1)
|
light red spot
|
|
C3...C8
|
-
|
-
|
3.391
|
-
|
(2)
|
light red spot
|
|
C13―H13A…O1A
|
0.971
|
2.305
|
3.276
|
171.46
|
(3)
|
dark red spot
|
|
C6―H6…O15
|
0.930
|
2.626
|
3.375
|
138.00
|
(4)
|
light red spot
|
|
H3―C3...H16B
|
0.930
|
2.854
|
3.028
|
101.73
|
(5)
|
light red spot
|
|
[(MnIII(HL)2Cl] (3)
|
|
C2…C17
|
-
|
-
|
3.255
|
-
|
(1)
|
dark red spot
|
|
C4―H4…C11
|
0.930
|
2.847
|
3.638
|
143.66
|
(2)
|
light red spot
|
|
C6―H6…Cl
|
0.930
|
2.856
|
3.694
|
150.43
|
(3)
|
dark red spot
|
|
[CoIII(HL)3] (4)
|
|
H5A…H16C
|
-
|
2.277
|
-
|
-
|
(1)
|
light red spot
|
|
C2A …C4C
|
-
|
-
|
3.380
|
-
|
(2)
|
light red spot
|
|
C7A―H7A…C8B
|
0.950
|
2.875
|
3.630
|
137.19
|
(3)
|
light red spot
|
|
C7A―H7A…C9B
|
0.950
|
2.777
|
3.584
|
143.28
|
(4)
|
light red spot
|
|
C14D―H14D…O15A
|
0.990
|
2.548
|
3.493
|
159.51
|
(5)
|
light red spot
|
|
H16F…H10B
|
-
|
2.102
|
-
|
-
|
(6)
|
dark red spot
|
|
C2C …H16A
|
-
|
-
|
3.235
|
-
|
(7)
|
light red spot
|
|
C16C―H16C…C4B
|
0.980
|
2.841
|
3.629
|
137.97
|
(8)
|
light red spot
|
|
C2C―H2C … O15C
|
0.950
|
2.647
|
3.258
|
122.57
|
(9)
|
light red spot
|
|
C3C―H3C … O15C
|
0.950
|
2.715
|
3.295
|
120.00
|
(10)
|
light red spot
|
Figures. 16
Table 3
Figure 17 shows a view of the Hirshfeld surface calculated by the shape index function. As it was predicted in this figure, similar to the studied complexes, the presence of π...π and C–H...π interactions with the special features of “blue- red triangle” (for π…π) and “red π-holes” (for C–H…π) were visible [22, 40].
Figure. 17
By studying the 2D fingerprint diagrams in Fig. 18, it was observed that the contribution of H···H exchange shown in the center of this diagram shows the greatest participation in the formation of the [CoIII(HL)3] complex (4) (it occupies about 61.2% of the Hirshfeld surface). In this complex, C–H⋯π interactions arise from C⋯H/H…C contact C…H/H…C (No: 2) and occupy a part of this diagram, which is about 27.2% of the diagram. covers (di + de < 2.8). In addition, O···H/H····O (No: 3) contacts are observed in this complex (exchanges (8.2%) (di + de < 2.6)). In addition, several interactions with a lower ratio contribute to the stability of the crystal system, such as: (C...C (3.2%), N...H (0.1%) and C...O (0.1%) [22, 40].
More information obtained from the 2D fingerprint graph for all four crystal structures is summarized in Table 4.
Based on the results obtained from the 2D fingerprint graph it is deduced that more than one half of the surface corresponds to H…H contacts for all four complexes. Some of these contacts seem as a tip on the diagonal diagram providing short exchanges of H...H with values di + de < 2.2 Å, for all complexes, i.e. less than 2 × the van der Waals radius of hydrogen atoms. For all complexes, the H…O/O…H contribution covering strong C―H…O hydrogen bonds is marked by two sharp peaks providing the short contacts with the H…O/O…H distances less than the sum of the van der Waals radii of hydrogen and oxygen atoms (≈ 2.7 Å) including the values of di+ de ≈ 1.1–2.5 Å and 2 Å (for 1 and 2), 2.7 Å (for 3) and 2.5 Å (for 4). In complex (3), small spikes on the fingerprint graph can be seen which are associated to the H…Cl/Cl…H contacts related to the C―H…Cl hydrogen bond interactions. Some of these contacts provide the H…Cl/Cl…H short contacts with values of di+ de ≈ 2.7 Å, i.e. slightly less than the sum of the van der Waals radii of hydrogen and chlorine atoms (≈ 2.8 Å). In the case of the crystal structures, the study of the FPs displays that only three H...H, C…H/H…C and H...O/O...H interactions play an effective role in the crystal packing with significant contributions as have been discussed.
Table 4
The information obtained from the 2D fingerprint graph with a larger contribution interaction for the complexes.
|
Interactions
|
Percentage of participation
|
di
|
de
|
di+de
|
|
[ZnII(HL)2CH3OH] (1)
|
|
H…H
|
54.5%
|
1.1–2.7
|
1.1–2.5
|
2.2
|
|
C…H
|
35.7%
|
1.6–2.7
|
1.6–2.5
|
2.6
|
|
O…H
|
8.9%
|
1.1–2.4
|
1.1–2.4
|
1.1–2.5
|
|
[ZnII(HL)2] (2)
|
|
H…H
|
53.6%
|
1.1–2.4
|
1.1–2.4
|
2.2
|
|
C…H
|
32.3%
|
1.7–2.5
|
1.7–2.4
|
2.8
|
|
O…H
|
11.3%
|
1.3–2.4
|
1.3–2.4
|
2.0
|
|
[(MnIII(HL)2Cl] (3)
|
|
H…H
|
46.6%
|
1.1–2.5
|
1.1–2.6
|
2.2
|
|
C…H
|
33.6%
|
1.7–2.5
|
1.7–2.6
|
2.8
|
|
O…H
|
8.9%
|
1.5–2.5
|
1.5–2.4
|
2.7
|
|
[CoIII(HL)3] (4)
|
|
H…H
|
61.2%
|
0.9–2.6
|
0.9–2.6
|
1.8
|
|
C...H
|
27.2%
|
1.6–2.7
|
1.6–2.6
|
2.7
|
|
|
O…H
|
8.2%
|
1.4–2.5
|
1.4–2.5
|
2.5
|
|
Table 4
Figure. 18
3.7. Computational Chemistry and Density functional theory (DFT) calculations
3.7.1. UV-Vis spectrum ([ZnII(HL)2 CH3OH] (1), [ZnII(HL)2] (2), [(MnIII(HL)2) Cl] (3) and [CoIII(HL)3] (4) complexes)
electronic transitions of studied complexes 1 to 4 in acetonitrile solvent were calculated using computational chemistry at the B3LYP/6-311G level. The calculations performed using theoretical studies were compared with the experimental data of this spectrum (Figure S4 in the experimental section). In the electron transfer spectrum of experimental data, intra-ligand transfer (C = N) showed absorption bands in the region of 304–344, 306–330 and 275–322 nm for complexes (1, 2), (3) and (4), respectively. In comparison with the spectrum obtained from the theoretical studies, the same regions show a lower absorption band for complexes 1 to 2, 292–301 (for complex (1)) and 305–313 (for complex (2)), and a higher absorption band for complexes 3 to 4 around 371–387 (for complex (3)) and 384–423 (for complex (4)). Moreover, charge transfer is assigned in the regions of 403–418 nm (for complex (1, 2)) and 396–417 nm (for complex (3)). Computational chemistry electron transfer spectrum in this region, lower absorption band around 364–370 nm with energy transfer 3.419–3.277 eV (complex (1)), 359–373 nm with energy transfer 3.453–3.320 eV (complex (2)) and higher absorption band around 400–459 nm with energy transfer of 3.099–2.699 it was calculated. The d → d absorption band of the experimental spectrum of the complex (complex (3) and complex (4)) is around 484 and 675 nm and this electron transfer in computational chemistry has shown absorption bands at 760 and 605 nm with an energy transfer of 1.968 and 2.048 eV, respectively. According to the discussed results, it is concluded that computational chemistry correctly suggests the structure of all complexes. Also, the energy gap difference shows well the stability, hardness and reactivity of the complexes, the higher the energy gap difference, the higher the stability and chemical hardness and low reactivity of the complex [41, 42]. Therefore, for complex (1), the largest energy gap was calculated compared to other complexes ((2), (3)), as a result, this compound shows a higher chemical hardness, these results are further detailed in the analysis section and Frontier molecular orbital analysis will be discussed.
3.7.2. Frontier molecular orbital analysis
LUMO and HOMO molecular orbital images calculated at the B3LYP/6-311G level of theory for all four complexes 1 to 4 are shown in Fig. 19. The LUMO molecular orbitals in complexes (1 and 2) are mostly located on the naphthaldehyde ring, (O)2−hydroxy atoms and HC = N atoms, while the HOMO orbitals are mainly distributed on the zinc metal, HC = N and contains very little naphthaldehyde rings. For complex (3) LUMO molecular orbitals, the coverage of the orbitals is mostly on the central metal Mn, HC = N and also covers a small part of the naphthaldehyde ring, but, for the HOMO orbitals has been seen on the naphthaldehyde ring, the central metal, HC = N and the group amine. For complex (4), the LUMO molecular orbitals are scattered on two ligands including the naphthaldehyde ring, (O)2−hydroxy atoms and HC = N atoms and the metal center Co, but the HOMO orbitals are covered by one ligand and quantitative surface of naphtaldehyde rings, Co, HC = N.
In Table 5, the values of the calculated HOMO and LUMO energy levels for the complexes are respectively − 7.146 and − 5.747 eV for complex (1), -7.234 and − 5.782 eV for complex (2), -6.066 and − 5.449 eV for complex (3) and − 5.823 and − 5.514 eV for complex (4). Interesting results were collected by mapping frontier molecular orbitals (FMOs). The calculated LUMO values indicate the ability to accept electrons (nucleophilicity), while the obtained HOMO values determine the nature of the molecule to donate electrons (electrophilicity) [41, 43]. If the calculated LUMO energy value is lower, it indicates a high ability to accept electrons [41, 43]. By observing the results obtained in Table 5, it is concluded that complex (3) has the lowest LUMO energy compared to other complexes. Also, the energy difference (ΔE) of complex (4) is lower than complexes (1 > 2 > 3 > 4), as a result, metal ions as an active center has a stronger potential to accept electrons or it has electron donation. Therefore, the biological activity of the complexes was predicted as 4 > 3 > 2 > 1 [44]. According to the calculated energy of orbitals, it was used to check quantum parameters such as reactivity, electronegativity (χ), chemical potential (µ), chemical hardness (η), softness (S) [41, 45, 46]. The parameters were estimated using Eq. 5 − 1 respectively (Table 5).
EGAP = ELUMO - EHOMO (1)
χ = -1/2(ELUMO + EHOMO) (2)
µ= - χ (3)
η = 1/2(ELUMO − EHOMO) (4)
S =\(\frac{1}{2{\eta }}\) (5)
The information related to the mentioned parameters for complexes 1 to 4 is presented in Table 5. The higher the energy difference between the HOMO and LUMO orbitals, the less polar and chemically reactive the molecule is, and it is referred to as a hard molecule, while the lower the energy difference between the orbitals, it is known as a soft molecule. According to the obtained information of these parameters, complex (2) with a higher energy gap, HOMO and LUMO, has a more stable structure than complexes (1), (3) and (4) and acts as a harder molecule. Complex (1) has a greater energy gap difference than (3), indicating that complex (1) is harder than (3). Complex (4) has a lower energy gap with more polarity and greater reactivity than complexes 1 to 3 (4 > 3 > 2 > 1, soft molecule).
|
Table 5. Calculated global reactivity descriptors of the three complexes at the B3LYP/6-311G level.
|
|
|
Parameters
|
Complex(1)
|
complex (2)
|
complex (3)
|
complex (4)
|
|
|
ELUMO − EHOMO (eV)
|
1.452
|
1.399
|
0.617
|
0.309
|
|
|
ELUMO + EHOMO (eV)
|
-12.893
|
-13.016
|
-11.515
|
-11.337
|
|
|
Electronegativity (χ)
|
6.5085
|
6.444
|
5.758
|
5.670
|
|
|
Chemical potential (µ)
|
-6.508
|
-6.444
|
-5.758
|
-5.670
|
|
|
Chemical hardness (η)
|
0.726
|
0.702
|
0.308
|
0.154
|
|
|
Softness (S)
|
0.688
|
0.712
|
1.620
|
3.243
|
|
Figure 19.
Table 5
3.7.3. Molecular electrostatic potential
One of the useful parameters for characterizing electrophilic and nucleophilic sites and hydrogen bonding exchanges is the molecular electrostatic potential (MEP) [47]. MEP maps are a visual representation of the electronic density for the title complexes, and these calculations are considered to relate the molecular structure to its physicochemical properties [48]. The 3D molecular electrostatic potential maps for the studied complexes are presented in Figs. 20, and the positive and negative locations of these complexes are colored with the electrostatic potential values increasing from red to blue. According to all four MEP schemes, it can be understood that oxygen atoms and Cl for complex (3) with red color have a negative electrostatic potential and strong repulsion and are candidates for electrophilic attack. The sites of strong attraction are the greatest possible regions with blue colors, which are observed near the C-H group, and central metal they provide highly reactive regions for nucleophilic attack. These locations are also presented for hydrogen bonding due to the existence of greatly electronegative atoms C, Cl and O. We were able to show the correctness of results MEP maps with the help of Hirshfeld surface analysis.
Figures 20.
3.8. Results from molecular docking studies
molecular docking was used to investigate the inhibitory potential of the [ZnII(HL)2CH3OH] (1), [ZnII(HL)2] (2) [(MnIII(HL)2) Cl] (3) and [CoIII(HL)3] (4) complexes to the target proteins (binding affinity to protein B-cell lymphorna (BCL-2, PDB ID: 4LXD)), which are important proteins involved in the cancer cell binding mechanism. By binding simulation, it was done by choosing the best state of the complexes towards the receptors with the highest negative binding energy. In this part, the interaction of complex (1–4) with an important molecular target in cancer treatment was done with the help of molecular simulation method [45, 46, 49–51]. By blocking ATP kinase, energy does not reach the cancer cells and this causes the cancer cells to die. Since cancer cells are able to absorb more energy, healthy cells receive less energy. Therefore, one of the inhibitory factors of cancer cells is ATP kinase, and in this section we used the PDB: 4LXD protein to study all four complexes [52, 53].
3.8.1. Results from molecular docking studies [Zn II (HL) 2 CH 3 OH] (1), [Zn II (HL) 2 ] (2), [(MnIII(HL)2)Cl] (3) and [CoIII(HL)3] (4) complexes
Two-dimensional and three-dimensional simulation models of the studied complexes are presented in Figs. 21–24 (a and b). First, the optimization of the studied structures was done, then, by performing the docking process, the complexes (1–4) were connected to the active site of the protein and the total energy of the system was calculated. In the case of the target protein 4LXD, the best ranking was obtained with a calculated total energy of the system − 27.48, -47.75, -64.66 and − 49.93 kcal/mol for complexes 1 to 4, respectively. By calculating the negative energies for the studied complexes, it is proved that the interaction of these complexes with 4LXD is able to disrupt the activity of this protein, therefore, these complexes can be suggested as inhibitors in cancer treatment. The results showed that the complexes 1 to 4 were able to create appropriate interactions with the active binding sites of proteins. Also, by forming various bonds, they showed that these structures inhibit the biochemical processes of protein 4LXD. As a result, the total system energy of complex (3) was lower than the total system energy of other studied complexes (1 < 2 < 4 < 3). In the Hirshfeld surface analysis study, complex (3) showed a different hydrogen bond due to the presence of chlorine atom in the structure compared to other complexes, due to this type of interaction in the crystal structure, molecular docking has shown a more favorable result. In addition, all the atoms described and involved in the intermolecular interactions of the Hirshfeld surface analysis are similarly involved in the molecular binding in the interaction between the protein and the receptor. The interaction of the active site amino acid of the receptor with the most potent inhibitor is briefly listed in Table 6 for all four complexes. In these complexes, proteins are able to penetrate well into the cavity of the active site of the receptors and cause the formation of more amino acids around the said complexes and cause non-covalent interactions and negative total energy of the system. Also due to the crystal structures of the complexes and the type of interaction in it and their favorable orientation in the active position of the receptors caused the formation of a favorable interaction with amino acids and increased the inhibitory power of this complex, complex (3) with the highest inhibitory activity against cancer protein (4LXD), with active amino acid residues the following interacted: PHE A:101,TYR A:105, ASP A:108, GLU A:142, ARG A:143, ALA A:146, PHE A:150, VAL A:153 and MET A:154, etc. through
|
Table 6. Amino acid interaction of the active site of the receptor with the strongest inhibitor.
|
|
|
Combined receptor
|
Total energy
|
Surrounding amino acids
|
|
[ZnII (HL)2CH3OH] (1)
|
|
|
(ID: 4LXD)
|
-27.48
|
PHE A:101, TYR A:105, ASP A:108, PHE A:109, GLU A:111, MET A:112, SER A:113, VAL A:130, LEU A:134, ARG A:136, ARG A:143, ALA A:146, GLU A:149, PHE A:150, VAL A:153.
|
|
|
[ZnII (HL)2] (2)
|
|
|
(ID: 4LXD)
|
-47.75
|
PHE A:101, SER A:102, TYR A:105, ASP A:108, PHE A:109, MET A:112, SER A:113, GLU A:116, VAL A:130, LEU A:134, GLU A:142, ARG A:143, VAL A:145, ALA A: 146, PHE A:147, GLU A:149, PHE A:150, VAL A:153, MET A:154, GLU A:157.
|
|
|
[MnIII (HL)2Cl] (3)
|
|
|
(ID: 4LXD)
|
-64.66
|
PHE A:101, SER A:102, TYR A:105, ASP A:108, PHE A:109, MET A:112, VAL A:130, GLU A:133, LEU A:134, ASN A:140, GLU A:142, ARG A:143, ALA A: 146, GLU A:149, PHE A:150, VAL A:153, MET A:154.
|
|
|
[CoIII (HL)3](4)
|
|
|
(ID: 4LXD)
|
-49.93
|
PHE A:101, SER A:102, TYR A:105, ARG A:106, ASP A:108, PHE A:109, GLU A:111, MET A:112, GLN A:115, VAL A:130, VAL A:131, LEU A:134, GLU A:142, ARG A:143, ALA A: 146, PHE A:147, GLU A:149, PHE A:150, VAL A:153.
|
|
Figures. 21–24
Table 6
{kind=link}