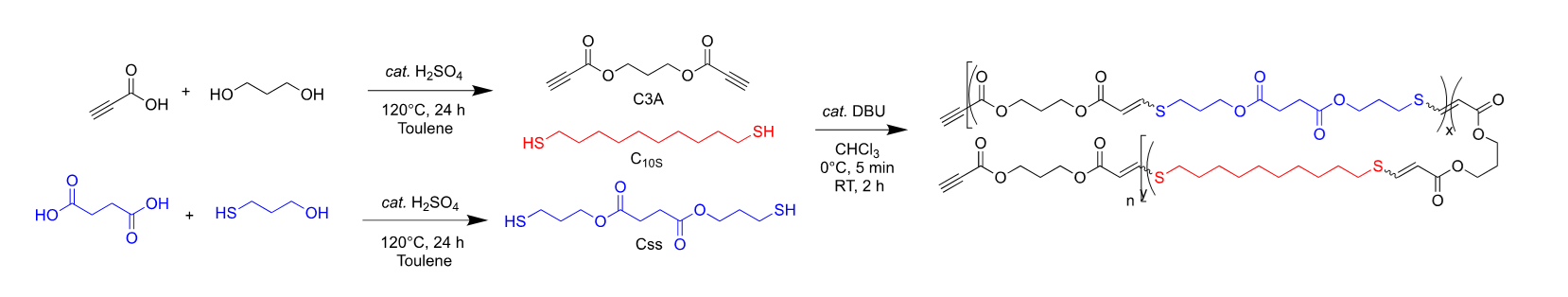
The copolyester library for the degradable barrier films was synthesized via a stereocontrolled thiol-yne polymerization. 18, 20–23 These polymerizations feature well-defined cis: trans ratios controlled by the nature of the organobase catalyst, solvent polarity (which influences the Kd between the organo-base and the propriolate monomer), and the reaction temperature. Previous work has shown an empirical correlation between higher cis content and increased crystallinity.18 Therefore, to obtain relatively high (~ 80%) cis stereochemistry, the reaction temperatures are maintained near 0°C (via a sodium chloride ice bath) with catalytic 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) in chloroform as outlined in Scheme 1.
The alkyne (C3A) and the succinate-based (Css) monomers were synthesized via Fisher esterification reactions and purified via column chromatography. The commercially available 1,10-decanedithiol (C10S), was introduced to increase the hydrophobicity of the resulting copolyester polymers. Each of the three monomers and DBU were distilled prior to polymerization. Before quenching the catalyst with BHT, a slight excess of C3A was introduced at the end of the polymerization to cap terminating thiol moieties which serves to avoid post-polymerization reactions. The resulting polymers were precipitated directly from the reaction mixture in diethyl ether yielding polymers of moderate molecular mass (55–95 kDa) with molar mass distributions (ĐM) near 2 as expected for step-growth polymerizations (Table 1). Thermo-mechanical characterization of these copolymers revealed that melting temperature (Tm) increased with decreasing Css stoichiometry. At high Css stoichiometry (60–100 mol% Css) the absence of a Tm or Tc is likely due to the ester moieties disrupting polymer packing to the extent that the material is no longer semi-crystalline. The Young’s moduli (E0) and ultimate tensile strengths (UTS) also decrease proportionally, accompanied by increases in strain at break, with increasing Css stoichiometry which is expected with the observed trend in crystallinity. The degradation temperature (Td) exceeded 340°C across all the copolymer compositions which affords a significant thermal processing window due to the wide gap between the Td and Tm.
Table 1
Summary of the Thermomechanical Properties of the Css copolymer Library.
| %Css | Mw (kDa) | ĐM | Tg (°C) | Tm (°C) | Tc (°C) | Td (°C) | E0 (MPa) | ebreak (%) | UTS (MPa) |
| 0 | 72.8 | 2.2 | -7 | 112 | 61 | 351 | 65.8 ± 0.6 | 1457 ± 112 | 32.2 ± 3.9 |
| 10 | 81.3 | 2.5 | -10 | 102 | 54 | 354 | 105.2 ± 1.4 | 1105 ± 67 | 26 ± 0.1 |
| 15 | 83 | 2.7 | -10 | 98 | 40 | 357 | 93.1 ± 2.1 | 1384 ± 108 | 25 ± 0.5 |
| 20 | 68.6 | 2.1 | -7 | 99 | 30 | 348 | 57.2 ± 4.4 | 1700 ± 31 | 40.5 ± 2.2 |
| 25 | 81.0 | 2.3 | -9 | 93 | 26 | 359 | 67.2 ± 1.7 | 1506 ± 37 | 25 ± 3.4 |
| 30 | 60.4 | 2.1 | -7 | 89 | 19 | 349 | 40.9 ± 3.5 | 1638 ± 33 | 28.3 ± 1.2 |
| 40 | 57.0 | 2.1 | -7 | 83 | 13 | 349 | 36.2 ± 2.0 | 2110 ± 50 | 36.2 ± 4.1 |
| 50 | 94.9 | 2.1 | -10 | 74 | 22 | 348 | 36.2 ± 2.0 | 1943 ± 155 | 37.4 ± 4.5 |
| 60 | 71.7 | 2.5 | -12 | - | - | 342 | 12.0 ± 1.4 | 2077 ± 78 | 20.5 ± 0.6 |
| 100 | 86.8 | 1.4 | -12 | - | - | 358 | 4.6 ± 0.3 | 1886 ± 147 | 8.4 ± 1.9 |
% Css: % incorporation of the Css monomer, Mw: Weight average molecular mass, ĐM: Molar Mass Distribution, Tg: Glass transition temperature, Tm: Melting temperature, Tc: Crystallization temperature, Td: Degradation temperature, aE0: Young’s modulus, aebreak: elongation at break, aUTS: ultimate tensile strength. a Determined from 3 or more replicates.
A series of differential scanning calorimetry (DSC) experiments with constant heating and cooling rate (10°C/min) were conducted to assess the influence of Css stoichiometry on the thermal transitions (Fig. 2A and 2B). While the glass transition temperatures, Tg, of the series of polymers was nearly constant (~ -7 to -12 °C), the melt transition temperatures, Tm, of the polymers decreased proportionally from 0% Css (112 °C) to 50% Css (74 °C). The crystalline behavior and the presence of a Tm was not observable beyond 50% Css incorporation after the first heating cycle. The crystallization temperature, Tc, also decreased proportionally from 0% Css (61 °C) to 40% Css (13 °C).
The existing correlation between crystallinity and barrier properties necessitated deeper investigation into the crystalline properties of the Css copolymers (15–25% Css).14, 24 The 15–25% Css stoichiometries exhibit similar wide-angle x-ray scattering (WAXS) diffraction patterns which is indicative that they possess similar crystalline structures (Fig. 2C). Xc and mean crystallite size were approximately the same across the investigated copolymers (15–25% Css) and the observed 2D scattering patterns in SAXS/WAXS are consistent with polycrystalline materials exhibiting many random modes of orientation (Fig. 2E). Each incorporation also demonstrated moderate to high water contact angles consistent with hydrophobic materials and improved barrier properties (Fig. 2E).27, 28
The properties of semi-crystalline polymers are strongly affected by their processing conditions.25, 26 Thus, DSC experiments with varied cooling rates (1 to 12°C/min) were conducted to observe how cooling rate after processing may affect polymer properties. These cooling rate experiments demonstrated that the crystalline properties of these polymers varied dramatically with cooling rate where slower cooling rates gave more unimodal, higher temperature crystallization temperatures, Tc (Fig. 3A). Across all of the Css polymer stoichiometries, low Tgs (sub-ambient) are observed which could partially explain the observed dependence in crystallinity on cooling rate as polymer systems demonstrate higher dynamic mobility above the Tg.29 This was further supported by the observation of very thin lamellar crystals which suggests that these structures are unstable (Fig. 2E).30, 31 Despite this, the Xc did not appear to change significantly when the polymer was stored at ambient temperature for several weeks (\({\chi }_{c,fresh20\%Css}=\) 23.1% vs. \({\chi }_{c,aged20\%Css}=\) 23.0%) though the mean crystallite size decreased approximately 14 Å between the freshly cast and aged, slow cooled 20% Css substrates (176 Å and 162 Å respectively).
Scanning electron microscopy (SEM) imaging of 20% Css films shed light on the differences in film surface quality following several thermal processing methods. The samples prepared by controlled cooling methods via DSC or which were blade-coated appeared more uniform with fewer observable defects in comparison to the fast-cooled compression-molded samples (Fig. 3D-F). This could be due to the observed dependence in crystallinity on cooling rate or differences in shrinkage.32 The slow-cooled sample also demonstrated very low surface roughness compared to commercially available polyimide films as determined by atomic force microscopy (RMS roughness = 9.7 ± 3.1 nm for slow-cooled 20% Css vs. 50000 ± 68300 nm for polyimide film). Low surface roughness is important for consistent barrier performance as non-smooth surfaces can exhibit thinner regions which fail before the rest of the film.
Wide-angle X-ray scattering (WAXS) diffraction patterns of 20% Css also showed that the processing method affected the homogeneity and size of the observed features between 13.6–16.6 Å (Fig. 3B). The larger feature predominated, and the extent of crystallinity (\({\chi }_{c}\)) was greater following longer cooling times (\({\chi }_{c,fastcooling}=\) 15.6% vs. \({\chi }_{c,slowcooling}=\) 23.1%). Variable temperature WAXS (VT-WAXS) experiments showed crystal growth in both slow (stepped) and fast-cooling conditions that resulted in a difference in the size and number of crystalline features. The cooling rate appears to affect the short-range order of amorphous compositions like the 60% Css copolymer as seen in both VT-WAXS and variable cooling rate DSC experiments (Figure S27).
Examination of Css crystallinity provides insight into ideal processing methods which maximize Xc and surface uniformity but cannot be used to draw direct conclusions about the barrier properties of the polymers. Dynamic vapor sorption (DVS) experiments with the 20% Css copolymer processed by three different methods confirmed that processing method considerably affects the rate and amount of water absorption (Fig. 3B). While blade-coating appeared to reduce the surface defects observed by SEM (Fig. 3C), it also showed reduced crystallinity when compared to the thermally processed samples. Overall, the slow-cooled melt-pressed sample showed the greatest crystallinity and barrier effect.
Dynamic vapor sorption experiments with a subset of the synthesized copolymers demonstrated that succinate stoichiometry directly trends with water absorption until the 20% Css copolymer (Fig. 4B). Notably, each of the tested copolymers demonstrated slower rates of water absorption (shallower slope) than poly(ethylene terephthalate) (PET) thin films.
A subset of Css copolymer were used to encapsulate serpentine magnesium traces at 37°C against the perpendicular diffusion of phosphate buffered saline in the experimental setup shown in Fig. 4A meant to mimic mg-based transient systems. The performance of these copolymers shows a similar trend in barrier performance to that of the DVS experiments, where barrier ability appears to increase with decreasing succinate stoichiometry. Remarkably, the 20% Css copolymer was able to extend the lifetime of these model devices over 40 days – making this composition a promising candidate for translational Mg-based implantable sensors. While the measurements are not directly on the implanted devices and controlling the thermal properties during the device fabrication will certainly need further optimization, the change in resistance over time is significantly less than other copolymer stoichiometries and the timelines are approaching clinical and commercial relevance. This surrogate system also exhibited some variability in performance which is likely due to shrinkage of the epoxy during curing that leads to microdefects in the tested films.
Molecular dynamics simulations of a subset of Css stoichiometries provided insight into why the 20% Css copolymer performed best despite exhibiting similar degrees of crystallinity to the neighboring stoichiometries. These simulations suggest that these copolymers self-assemble due to the differences in polarities of Css and C10s (Fig. 5). To quantify the relative polarity of the chain segments, the partial charge distributions were calculated for three monomer sequences by using B3LYP 6-31G* (d,p) basis set for AM1 optimized structures implemented in Gaussian 09. The results of this calculations show that CSS has the highest level of polarity (the magnitude of variation of partial charges between groups of atoms) among all three monomer sequences (Figure S31). The alkyl chain in the central segment in C10S is symmetric and appears to be fully screened. Therefore, the order of the polarity for three repeats is CSS>C3A>C10S. Considering the regularity of the sequence distribution in a copolymer chain and their relative polarity, two neighboring monomer sequences C3A-CSS and C3A-C10S pairs were treated as coarse-grained beads of type A (blue, more polar) and B (red, less polar) respectively (Fig. 6A).
To make the system as simple as possible while still obtaining meaningful information, simulations were performed on copolymer chains with n = 1 in a melt. The list of the studied systems is summarized in Table S1. Figure 5B shows snapshots of the simulation box for copolymers with different composition which corresponding scattering functions are plotted in Fig. 5C. The observed trend of scattering intensity with copolymer composition is consistent with X-ray diffraction data shown in Fig. 5D. Thus, the nonmonotonic dependence of intensity could be explained by transformation of the self-assembled structures from spheres to intertwined cylinders (gyroid-like morphology) of A-blocks. The additional scattering functions and snapshots of the simulation box illustrating morphology transformation with copolymer composition are shown in Figure S32. This difference in assembled structure could provide insight into superior barrier performance exhibited by the 20% Css copolymer with respect to other stoichiometries.
Implantable devices, particularly those which are intended to degrade in vivo, must be verified for tissue compatibility. Disks of a subset of the synthesized copolymers with 200 µm thickness along with polytetrafluoroethylene (PTFE) controls, were implanted subcutaneously for 10 weeks to observe their degradation behavior and tissue inflammatory responses in vivo. PTFE is a suitable control as it is widely used for medical device fabrication, does not degrade over time, shows excellent barrier properties, and provides corrosion resistance.33
No gross inflammation was evident from macroscopic observations postmortem for any of the polymers. Across all Css copolymer groups, fibrous capsule formation was found to be < 100 µm and uncalcified, which indicated that no severe inflammatory response occurred towards the copolymer and is within the range of an accepted response for an implantable material (Fig. 6). The absence of lymphoid cell aggregation or cuffing of the surrounding blood vessels suggested no immunogenicity. Furthermore, there was no evidence of necrosis noted and multinucleated giant cells were either sparse or lacking in the capsule walls. Ultimately, comparison of the tissue-material interfacial region between the Css copolymers and PTFE controls indicated the implanted polymers were biocompatible and biostable (Fig. 6).
Preliminary performance of the 20% Css copolymer in a prototype device (Fig. 7A-C) demonstrated the sensitivity of this system to physiological parameters. Using two different thicknesses, 50 and 100 mm, the devices demonstrated slow shifts in resistance values as seen previously in the surrogate testing. While additional efforts will be needed for interlayer adhesion optimization and controlling temperature to maximize crystallinity within the device barrier films, the sensors nearly double the functional lifetimes of the best performing wax barrier systems and are thin enough to be deformed by cerebral spinal pulsation impedance measurements.
{kind=link}