In this study, we performed the combined multi-omics analyses of the oral microbiome and metabolome of METH addicts under detoxification. Our results showed that even after six months of detoxification from METH, the oral microbiome and metabolome of these individuals were significantly different from those of healthy normal subjects, and these differences were characterized by molecular mechanisms underlying the toxic impairment and addiction of METH. Furthermore, our study identified a group of microbial markers associated with METH addiction, showing significant potential for accurate and effective detection of drugged driving.
METH enhances dopamine and norepinephrine levels in the synaptic gap, and long-term use of METH causes neurotoxicity in axon terminals [19]. These toxic effects can lead to not only neuropsychiatric disorders such as stroke and Parkinson's [20], but also adverse effects on the cerebrointestinal pumping and immune system [3]. The cardiovascular diseases are the leading cause of death in METH users, especially after overdose [21]. However, there are still insufficient studies on the molecular mechanisms of toxic damage and addiction of METH, leading to the inadequate treatment of METH detoxification based on psychotherapy with no specific medication available [6].
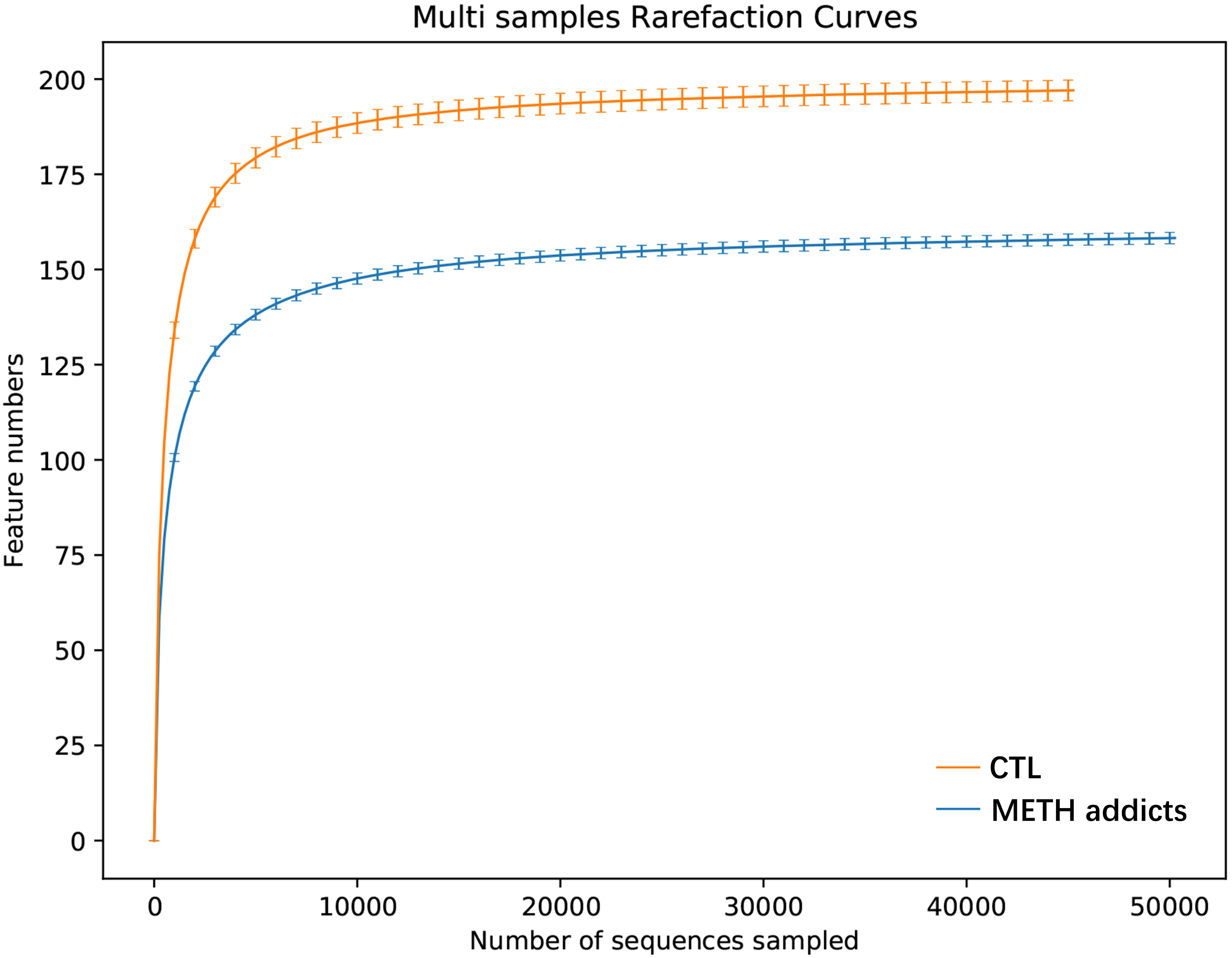
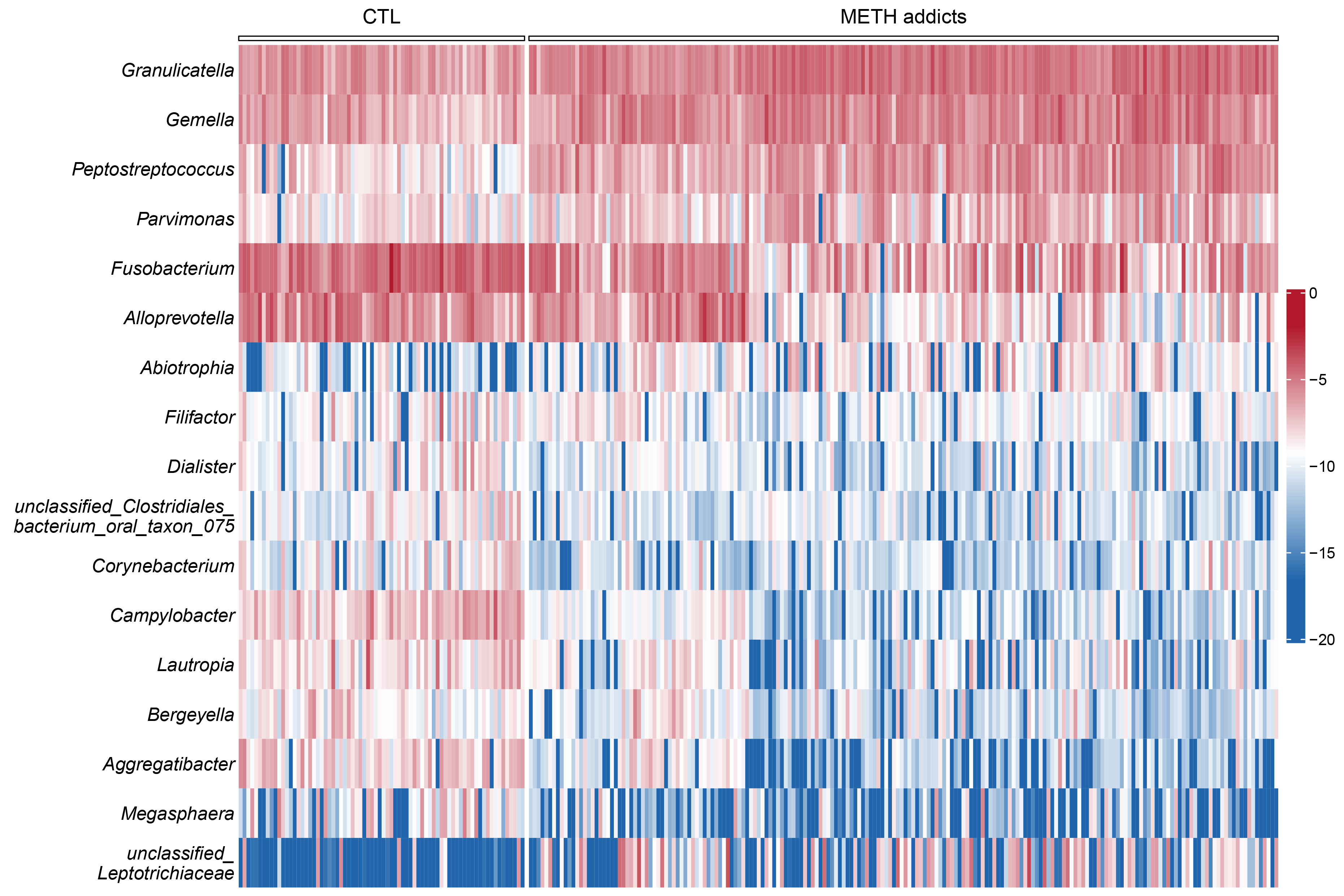
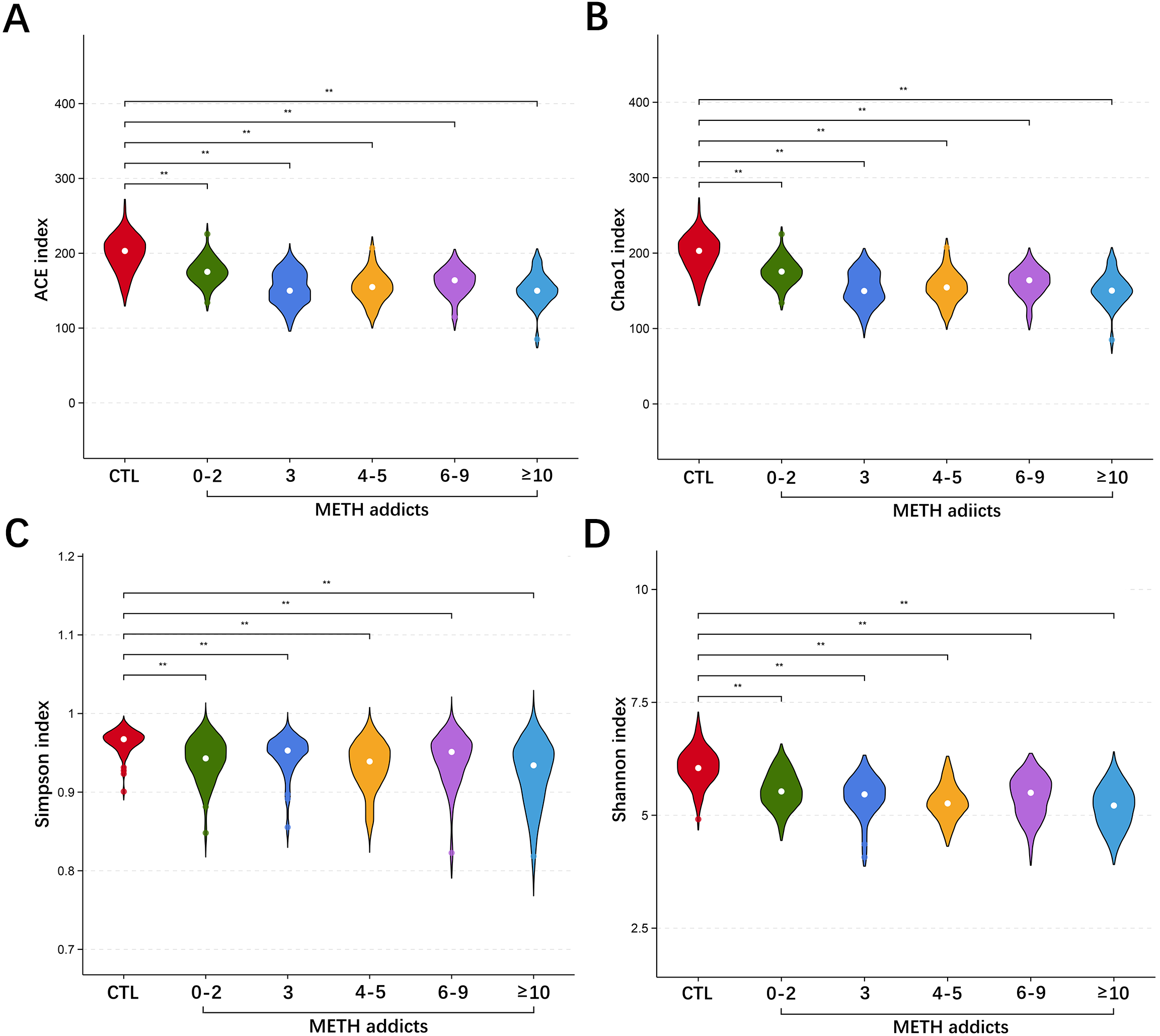
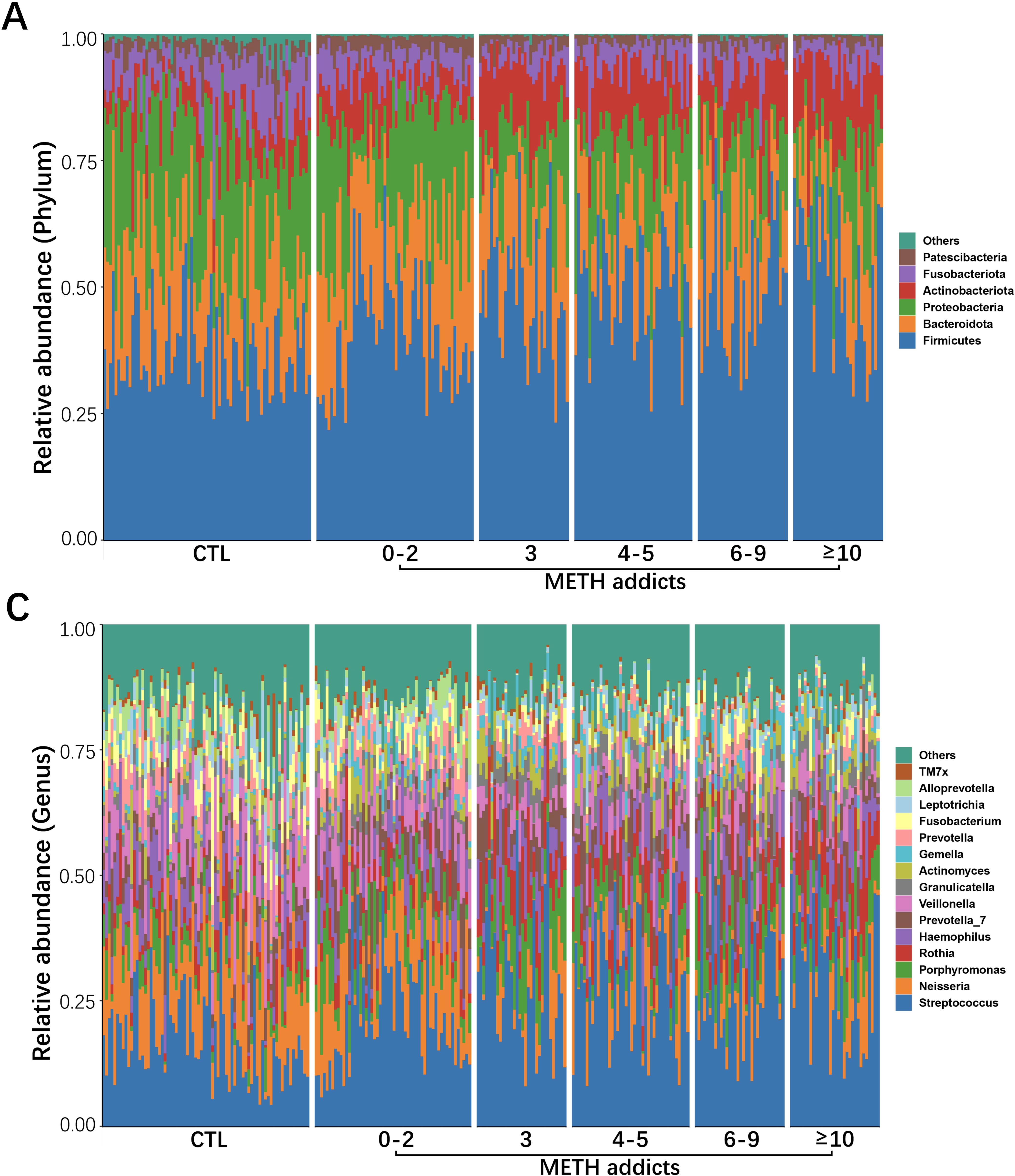
The oral cavity provides a highly heterogeneous ecological niche for microorganisms, and the damage to the oral microbial community caused by drug addiction is generally not permanent [9]. In our study, as one of the dominant phyla of oral microbiota observed in METH addicts, Firmicutes were revealed with significant changes of reduced alpha diversity, microbial community shifts, and alterations in the abundance of individual taxa. At the genus level, the alterations were observed in key microbiota probably involved in the mechanisms underlying the METH toxic injury. In the group of METH addicts, both Peptostreptococcus and Gemella were significantly increased, while both Campylobacter and Aggregatibacter were significantly decreased, compared to the CTL group.
Studies have shown that Peptostreptococcus was positively correlated with the cardiometabolic biomarker high-sensitivity C-reactive protein, which was involved in the cardiovascular diseases [22]. Furthermore, as a potential mediator of microbiome ecological dysregulation, Peptostreptococcu is enriched in the gut microbiota of patients with autoimmune diseases and ischemic stroke [23]. Gemella is also a part of the human oral microbiome. As the most common infection caused by Gemella, the infective endocarditis is also positively associated with the cardiovascular outcomes [25]. Furthermore, Gemella was also enriched in the oral microbiome of Parkinson's patients and smokers and was positively correlated with pro-inflammatory cytokines [26]. Studies have shown that catecholamines, norepinephrine, and dopamine promote the growth of Campylobacter in a strain-dependent manner and its pathogenicity [27]. It has been reported that Aggregatibacter may promote the development of neurological diseases with the Aggregatibacter aggregates detected in patients with Parkinson's disease [28].
Recently, Zhang et al. found that METH addiction caused a decrease in alpha diversity of the oral microbiota and an increase in alpha diversity during detoxification with Bacteroidetes identified as the dominant phylum and a group of five key microorganisms (i.e., Neisseria subflava, Haemophilus parainfluenzae, Fusobacterium periodonticum, Prevotella melaninogenica, and Veillonella dispar) involved in influencing the mechanisms regulating the METH addiction, providing high predictive accuracy for differentiating METH addicts based on a random forest classifier [9]. However, our study revealed different variations in oral microbiota, probably due to (1) the varied geographical sources of the samples, and (2) the length of detoxification, the smoking status, and the oral health of the participants, which could affect the compositions of the oral microbiota [29]. Considering that different microorganisms could perform similar functions [30], the molecular mechanisms underlying the toxic damage and addiction of METH cannot be fully explained from the microbial perspective alone.
To date, the growing evidence suggests that microorganisms perform different functions by the metabolites they produce, and the metabolites have become the important bridges between microorganisms and diseases in microbial pathogenesis [31]. Integrated analysis of multi-omics from microbiome and metabolome can provide clues to the mechanistic connections between microbiome and diseases [32]. Therefore, we performed a metabolomic analysis of the saliva samples. Our results showed that many differentially expressed metabolites in the group of METH addicts were associated with the metabolisms of amino acids, nucleotides, lipids, carbohydrates, cofactors, and vitamins. In particular, the metabolic pathways of tryptophan metabolism, lysine biosynthesis, purine metabolism, and steroid biosynthesis were enhanced in the group of METH addicts, while the metabolic pathways of porphyrin metabolism, glutathione metabolism, and the pentose phosphate were significantly reduced.
Currently, the tryptophan metabolic pathway is considered a major pathway connecting multiple systems and is closely associated with neuropsychiatric disorders [33]. It was found that 5-HT2A receptors were the key mediators of the addictive effects of METH, and tryptamine could inhibit the basal electrical activity of dopamine neurons by participating in tryptophan metabolism to produce 5-hydroxytryptamine (5-HT) [34]. Furthermore, METH has been reported to interact with the re-uptake of 5-HT, resulting in an acute increase in extracellular level of 5-HT, which, in addition to dopamine, is a key mechanism underlying the drug addiction [35]. Moreover, a recent study reported that the withdrawal syndrome following the termination of drug addiction is also associated with profound changes in 5-HT activity [36]. Leucyl-isoleucine is involved in leucine biosynthesis, and mutations in leucine-rich kinases are the common causes of Parkinson's disease, leading to dopamine neuron loss and motor dysfunction through abnormal increases in neuronal protein synthesis [37]. The (2R,3R)-3-Methylglutamyl-5-semialde-N6-lysine is an amino acid amide involved in lysine biosynthesis. Previous studies showed that trimethylation of lysine was enhanced in the voxel nuclei of the brain in a METH-induced behavioral sensitization model [38]. Furthermore, acetylation of lysine plays an important regulatory role in cardiovascular disease [39]. Both xanthine and inosine are involved in purine metabolism, while both purine and pyrimidine-related metabolites (e.g., xanthine base and adenosine 5'-monophosphate) are sensitive to METH addiction [40]. Studies have shown that purine metabolism is also closely associated with cardiovascular disease, metabolic syndrome, and chronic kidney disease [41]. The 3alpha,11beta,21-Trihydroxy-20-oxo-5beta-pregnan-18-al is involved in steroid hormone biosynthesis, while corticosterone exposure enhances METH-induced vascular damage, neuroinflammation, neuro degeneration, and lethality [42]. It has been reported that steroid hormones are involved in the pathogenesis of both Parkinson's disease and cardiovascular disease and are associated with the activation of pro-inflammatory mechanisms [43]. Glutathione helps maintain the normal immune system function, showing antioxidant effects to protect neurons from oxidative damage [44]. In addition, both the porphyrin metabolism and activation of the pentose phosphate pathway are associated with neuroprotection [45].
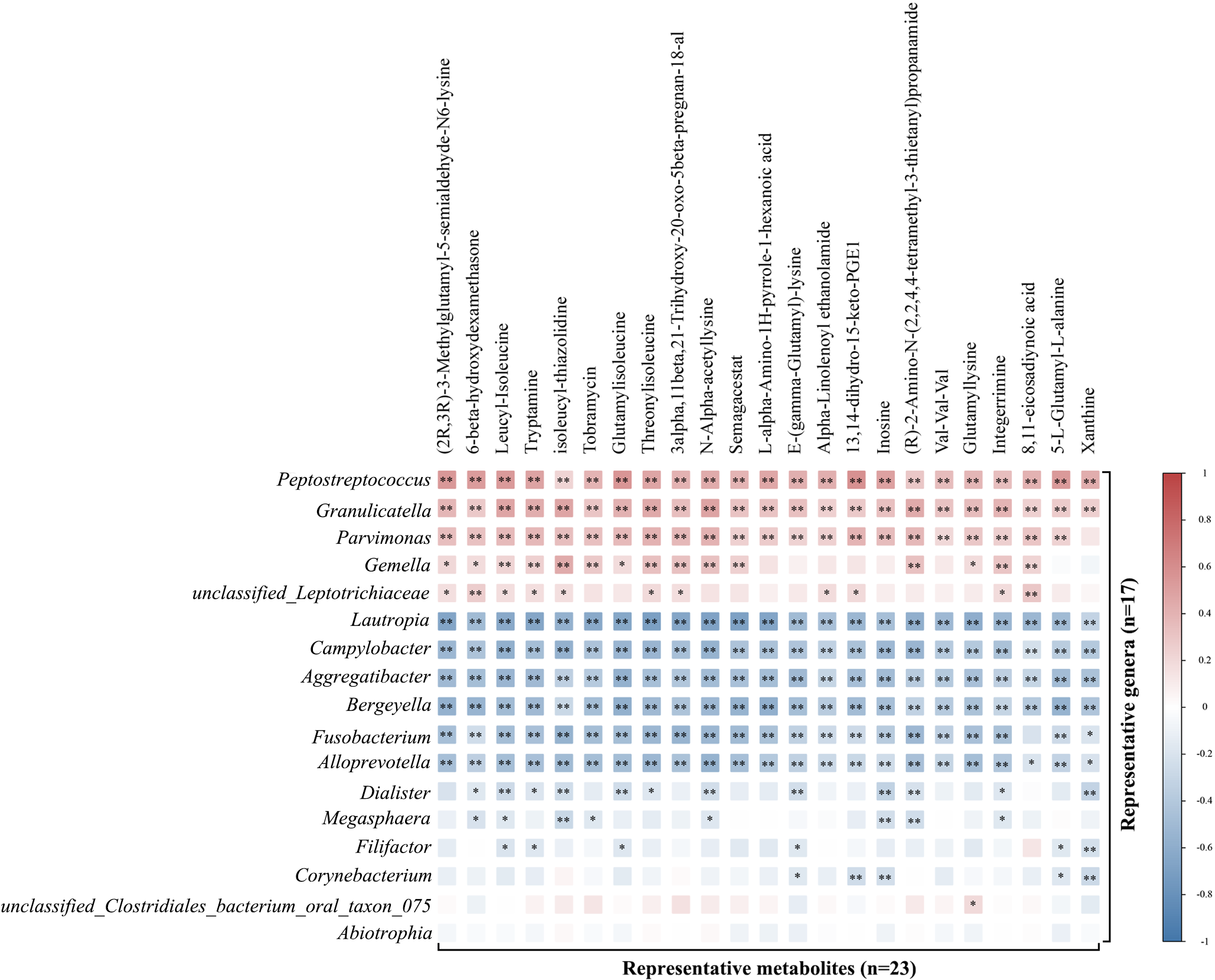
Based on the integrated multi-omics data, our study revealed strong associations between oral microbiota and metabolites in METH addicts. In particular, four key microorganisms, i.e., Peptostreptococcus, Gemella, Campylobacter, and Aggregatibacter, were significantly associated with six key metabolites, including tryptamine, leucyl-isoleucine, (2R,3R)-3-Methylglutamyl-5-semialde-N6-lysine, 3alpha,11beta,21-Trihydroxy-20-oxo-5beta-pregnan-18-al, xanthine, and inosine (Fig. 4A). These results were consistent with those previously reported, i.e., Peptostreptococcus, Gemella, and Campylobacter were involved in the pathogenesis of diabetes, depression, and oral cancer via the tryptophan metabolic pathway, respectively [47]. Furthermore, the association between Peptostreptococcus and steroid hormone synthesis as well as purine metabolism has been detected in gastric cancer and premature adrenal disease [50]. Therefore, it was hypothesized that these four key microorganisms (i.e., Peptostreptococcus, Gemella, Campylobacter, and Aggregatibacter) are involved in the molecular mechanisms regulating the toxic damage and addiction of METH via the metabolic pathways of tryptophan metabolism, lysine biosynthesis, purine metabolism, and steroid biosynthesis. In conclusion, our study demonstrated that METH disrupted the oral microbial ecological balance and it was difficult to recover the oral microbiota even after six months of detoxification, while the alterations in the compositions of oral microbiota and metabolites could be involved in the toxic damage and addiction process of METH.
The limitations of this study are noted. First, the female samples were underrepresented in the METH addicts. Second, most of the participants were smokers, while only the dietary habits of the participants were obtained through the questionnaire, potentially confounding the results of this study by the lifestyle and dietary characteristics of the participants. Finally, the lack of blood samples from participants in our study prevented further analysis of oral microbiota and metabolites with clinical indicators. Although our study revealed a functional connection between microbiome and metabolome, it could not define a cause and effect relationship. Further studies based on a more comprehensive sampling, i.e., with blood samples and dietary characteristics on the oral microbiome and metabolome, are needed to verify the findings revealed in our study.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}