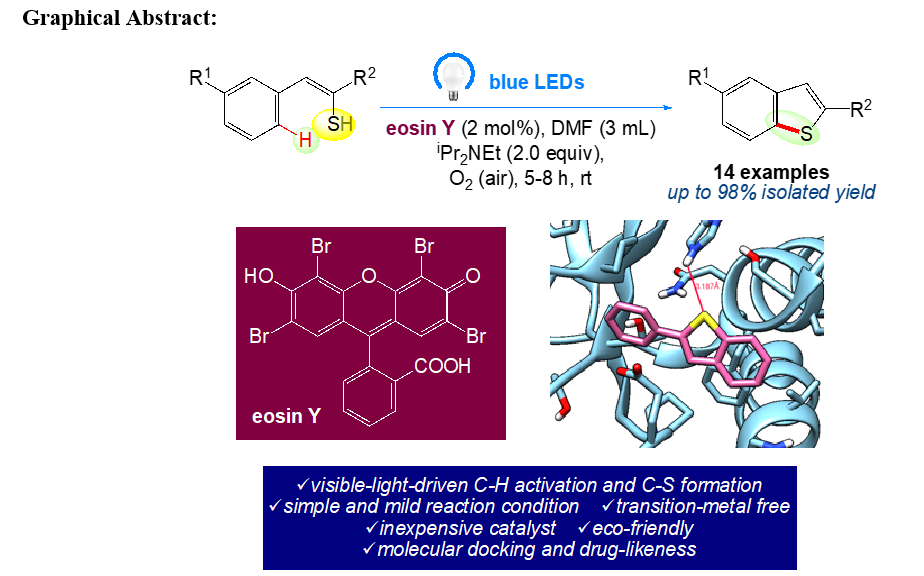
Visible light driven synthesis of substituted benzo[b]thiophenes from phenylethenethiol under an air atmosphere at room temperature is reported, using eosin Y as an organophotoredox catalyst by intramolecular C−S bond formation. This process accepts a wide range of functional groups tolerance and generates benzo[b]thiophenes under highly environmentally benign conditions via transition-metal-free organic photoredox catalysis.
Research Article
Visible-light photocatalysed Synthesis of Bioactive Benzo[b]thiophenes via Intramolecular C−S Bond Formation
https://doi.org/10.21203/rs.3.rs-2948452/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Substituted benzo[b]thiophenes are heterocycles of vital significance because of its occurrence in bioactive molecules [1–2], which include their use as HIV-1 reverse transcriptase inhibitors, antidepressants, and tubulin polymerization inhibitors [3–5]. These heterocycles also consist the basic structural unit present in commercial drugs, such as raloxifene [6–8] and arzoxifene [9–11], (selective estrogen receptor modulators) [12], Zileuton [13–14] (inhibitor of 5-lipoxygenase), Ipragliflozin [15–17] (type 2 diabetes), and Sertaconazole [18] (antifungal agent) (Fig. 1). Furthermore, benzo[b]thiophene-containing substances have been linked to antitubulin [19], antimitotic [20], urotensin-II receptors antagonistic [21], S1PR4 agonistic [22], anti- tubercular [23], and antinociceptive [24] activities in recent studies. Additionally, a lot of benzo[b]thiophenes may be used in the manufacturing of optoelectronic materials [25–30].
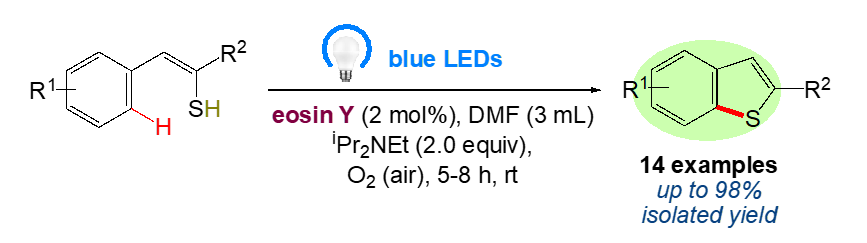
The use of UV or visible light to generate intramolecular C-S bonds has recently drawn more attention. Initially, the previously reported techniques for the synthesis of benzo[b]thiophene have been reported in literature due to their significance [31–39]. There are numerous prospects for the development of environmentally friendly and sustainable protocols for organic synthesis because solar energy (visible light) is a pure, easy-to-use, and limitless energy source [40–43]. Several trailblazing scientists [44, 45] have focused on the conversion of solar energy into chemical energy for chemical reactions and have also created a promising technique for using photoredox catalysts to initiate single electron transfer (SET) processes [46–48]. Recently, metal-free organic dyes such eosin Y, fluorescein, rose bengal, nile red, perylene, and rhodamine B have been employed as superior substitutes for transition metal photoredox catalysts, both economically and environmentally. In the current study, we focused on further exploring how a photoredox catalyst that emits visible light can be used to cyclize phenylethenethiol in an environmentally friendly manner. In keeping with our ongoing research into environmentally friendly synthetic methods [49–74], herein we proposed the application of a natural organic dye eosin Y, as photocatalyst using visible blue led to promote the transition-metal free chemical transformation under mild reaction conditions. (Scheme 1).
In order to work out the envisaged protocol, a key reaction was conducted with phenylethenethiol 1(a–n) in DMF containing 2 mol % of eosin Y under an air atmosphere (without air bubbling) by irradiation with blue light (blue LEDs, λmax = 467 nm) at rt. The reaction delivered the desired benzo[b]thiophenes 2(a–n) with moderate to excellent yield. (Table 1).
Table 1 Optimization of reaction conditionsa

|
Entry |
Photocatalyst |
Catalyst loading (mol%) |
Solvent |
Base |
Time (h) |
Yieldb (%) |
|---|---|---|---|---|---|---|
|
1 |
Eosin Y |
2 |
DMF |
iPr2Net |
5 |
92 |
|
2 |
Rose Bengal |
2 |
DMF |
iPr2Net |
5 |
86 |
|
3 |
Fluorescein |
2 |
DMF |
iPr2Net |
5 |
76 |
|
4 |
Benzophenone |
2 |
DMF |
iPr2Net |
5 |
71 |
|
5 |
Eosin Y |
2 |
MeOH |
DBU |
5 |
61 |
|
6 |
Eosin Y |
2 |
EtOH |
DABCO |
5 |
63 |
|
7 |
Eosin Y |
2 |
DMSO |
Et3N |
5 |
56 |
|
8 |
Eosin Y |
1 |
DMF |
iPr2Net |
5 |
62 |
|
9 |
Eosin Y |
3 |
DMF |
iPr2Net |
5 |
92 |
|
10 |
Eosin Y |
2 |
DMF |
iPr2Net |
5 |
42c |
|
11 |
Eosin Y |
2 |
DMF |
iPr2Net |
8 |
Traced |
|
12 |
- |
- |
DMF |
iPr2Net |
8 |
n.d.e |
aReaction conditions: phenylethenethiol (1.0 mmol), eosin Y (2.0 mol%), iPr2NEt (2.0 equiv.), DMF (3.0 mL), blue LEDs 3 W, irradiation under an air atmosphere at rt.
bIsolated yield of the product (2a).
cThe reaction was carried out using 20 W CFL (compact fluorescent lamp).
dReaction was performed in the dark.
eReaction was carried out without catalyst.
Following this experiment, a series of control experiments were performed, which indicates that an organic base is essential to give the desired product with high yield (92%) (Table 1, entry 1) and iPr2NEt was found to be the best base (Table 1, entry 1 versus 5, 6, 7). Next, the reaction conditions were optimized with respect to catalyst and eosin Y was found the best suitable catalyst. (Table 1, entry 1 versus 2, 3, 4). There was no product formation or it was formed in traces in the absence of any one of the reagents/ catalyst (Table 1, entries 11, 12). The reaction did not proceed satisfactorily when a household 20 W fluorescent lamp was used instead of blue LEDs (Table 1, entries 10 versus 1 These results establish that visible light, base, and photocatalyst all are essential for the reaction and supports this photocatalytic model of the reaction.
Next, the reaction conditions were optimized with respect to solvents and the catalyst used in the reaction. In all the tested solvents (DMF, DMSO, MeOH and EtOH) the yield of 2(a–n) was > 55% (Table 1), which indicates that the reaction is not very sensitive to reaction media. DMF was the best solvent in terms of the reaction time and yield (Table 1, entry 1), hence it was used throughout the synthesis. When the amount of the catalyst was decreased from 2 mol % to 1 mol %, the yield of 2(a–n) considerably reduced (Table 1, entry 8), but the use of 3 mol % of the catalyst did not affect the yield (Table 1, entry 9).
We next shifted our attention to exploring the adaptability of alternative substrates or anticipated reaction conditions, as we proceeded to search for the ideal reaction conditions for our model reaction (Table 2). The implementation of the present approach to a variety of phenylethenethiol including different substituents was investigated.
This clearly shows that the reaction is very mild and applicable to aryl and substituted aryl, tolerates considerable functional group variations like, NHCOMe, CO2Me, COMe, CHO, MeO, Me, Cl, Br and NO2 in the substrate 1(a–n), which results the desired product 2(a–n) in moderate to excellent yields (48–98%). However, phenylethenethiol with an electron-donating group on the aromatic ring appear to react faster and afford marginally higher yields in comparison to those bearing an electron withdrawing group.
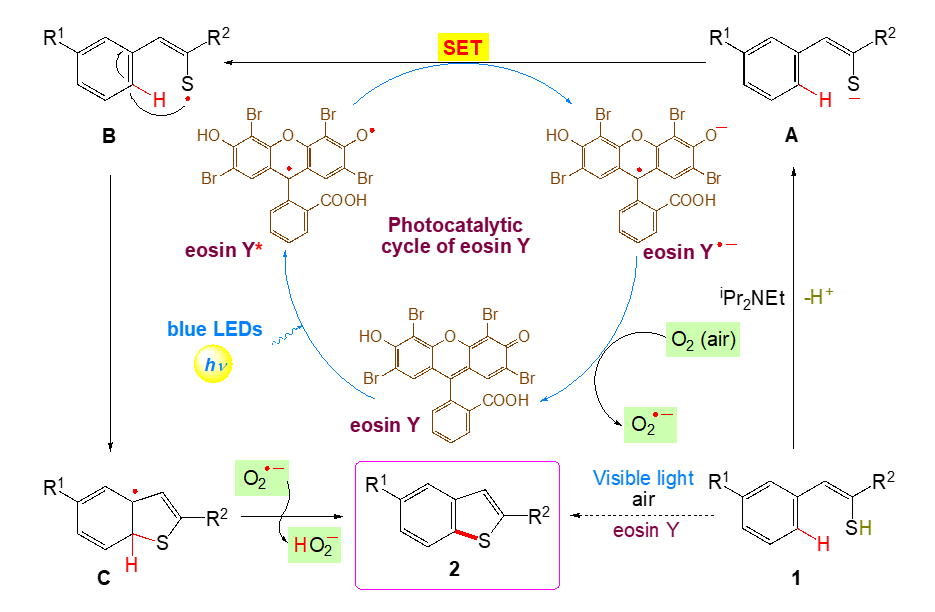
On the basis of the above observations and the literature precedents, a plausible mechanism involving photoredox catalysis for the oxidative cyclization of phenylethenethiol is depicted in Scheme 2.
On absorption of visible light, the organophotoredox catalyst eosin Y (EY) is excited to its singlet state 1EY* which through inter system crossing (ISC) comes to its more stable triplet state 3EY* and undergoes a single electron transfer (SET). 3EY* may undergo both reductive and oxidative quenching. [75–79] A SET from A to 3EY* generates thioacyl radical B, which undergoes intramolecular cyclization to form C followed by attack of O2• − to give the product 2, successively. The formation of superoxide radical anion O2• − during the reaction was confirmed by the detection of the resulting H2O2 using KI / starch indicator [80].
Experimental section
General information
Melting points were determined by open glass capillary method and are uncorrected. All chemicals used were reagent grade and were used as received. 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE DPX (400 MHz and 100 MHz) FT spectrometer in CDCl3 using TMS as an internal reference (chemical shift in δ, ppm).
General Procedure for the Synthesis of benzo[ b ]thiophenes 2(a–n)
A solution of a substituted phenylethenethiol 1(a–n) (1.0 mmol), eosin Y (2.0 mol %) and iPr2NEt (2.0 equiv.) were added and the mixture was irradiated with blue LEDs (3 W, λmax = 467 nm) with stirring under an air atmosphere at rt for 5–8 h. After completion of the reaction (monitored by TLC), water (5 mL) was added and the mixture was extracted with EtOAc (3 × 5 mL). The combined organic phase was dried over MgSO4, filtered and evaporated under reduced pressure. The resulting product was purified by silica gel column chromatography using a gradient mixture of hexane/ ethyl acetate as eluent to afford an analytically pure 2(a–n). All the products are known compounds and were characterized by the comparison of their spectral data with those reported in the literature.
Molecular Docking
Molecular docking is an accepted technique to identify bioactive compounds for drug development by calculating the binding affinity of the protein and ligand [81]. By using a computational technique called protein-ligand docking, it is possible to predict how and to what extent small compounds will bind to protein receptors. This molecular modelling technique [82–85] predicts the preferred orientation of one molecule to another when they are joined together to form a stable complex [86]. The pharmaceutical industry is well known for providing drugs development a high priority. By using online drug target prediction, also known as Swiss ADME-Target prediction, this compound, also referred to as a ligand, interacts with the specific protein that has been selected. Software Chimera 1.14 and Auto Dock Vina are used to dock 2-Phenylbenzo[b]thiophene molecule with 4FGL protein which belongs to oxidoreductase domain [87]. Table 3 provides a summary of the results, and Fig. 2 shows the best binding energy of -5.9 kcal/mol. A low binding energy value indicates that the compound is bioactive in nature and it indicates if the given ligand is suitable for the given proteins. The protein 4FGL contains 3 residues and the value of its Inhibition constant (KI) is 47.2.
|
Target Molecule |
2-Phenylbenzo[b]thiophene |
|---|---|
|
Protein (PDB ID) |
4FGL |
|
No. of Residues |
3 |
|
Bond Distance (A0) |
3.18 |
|
Inhibition constant (micromolar) |
47.2 |
|
Binding energy (Kcal mol− 1) |
-5.9 |
|
Reference RMSD (Å) |
10.87 |
Drug- likeness
The structural properties of the ligands in pharmaceutical study play a significant role to provide accurate and effective results for guidelines, also known as drug-likeness. In this technique, numbers of rules are used such as Veber rule, BBB rule, QED, Ghose filter, Lipinski's rule, MDDR-like rule and CMC-50 rules [88]. To identify whether a chemical molecule has the chemical and physical properties that would make it likely to be an orally active medicine in humans, Lipinski's rule [89] is a widely used filtering criteria. Efficiency is achieved when the 2-Phenylbenzo[b]thiophene and its derivatives are subjected to the drug-likeness criteria. Numerous activities are shown by these compounds. The vital ADME parameters, including the blood-brain barrier penetration (BBB) log kp, hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), topological polar surface area (TPSA), bioavailability score and molar refractivity (MR) are calculated for the standard compound and its derivatives and are shown in Table 4. It is recommended that both HBA and HBD readings should be below 10. The molecules in this particular case all have values that are less than 3. The highest value for TPSA should be 140 A2. For the target molecule and its derivatives, this ranges from 28.24 to 74.06. Table 4 shows that the bioavailability score of 2-Phenylbenzo[b]thiophene and its derivative is equal to 0.55 and that the skin's permeability (log Kp) is between − 3.97 and − 4.45. Additionally, all the target molecules have the relevant value of GI absorption. In addition, the molar refractivity should be around 40–130 [90]. The MR values of 2-Phenylbenzo[b]thiophene, 2-(4-nitrophenyl)benzo[b]thiophene and 5-methyl-2-(p-toly)benzo[b]thiophene are 67.26,76.08 and 77.19. According to the comparison described above, the title molecule, 2-Phenylbenzo[b]thiophene, and its derivatives are valuable building blocks that are commonly used in the development of a variety of substances with biological activity and medicines.
Table:4 ADME properties of 2-Phenylbenzo[b]thiophene and its derivative.
|
ADME properties |
2-Phenylbenzo-[b]thiophene |
2-(4-nitrophenyl)benzo-[b]thiophene |
5-methyl-2-(p-toly)benzo- [b]thiophen |
|---|---|---|---|
|
HBA |
0 |
2 |
0 |
|
HBD |
0 |
0 |
0 |
|
TPSA A2 |
28.24 |
74.06 |
28.24 |
|
MR |
67.26 |
76.08 |
77.19 |
|
GI absorption |
High |
High |
High |
|
BBB permeant |
Yes |
No |
Yes |
|
CPY1A2 inhibitor |
Yes |
Yes |
Yes |
|
Log kp (cm s− 1) |
-0.05 |
-4.45 |
-3.97 |
|
Lipinski violations |
0 |
0 |
1 |
|
Bioavailability score |
0.55 |
0.55 |
0.55 |
HBA- hydrogen bond acceptor, HBD- hydrogen bond donor, TPSA-topological polar surface area, MR- molar refractivity, GI- gastrointestinal, BBB- blood-brain barrier penetration and log kp- skin permeability.
In conclusion, we have developed visible light induced protocol for the synthesis of bioactive benzo[b]thiophenes, via C-S bond formation at room temperature. This method includes a novel organocatalysed protocol for the synthesis of benzo[b]thiophenes directly via cyclization of phenylethenethiol in a one-pot procedure by using inexpensive eosin Y as a powerful organophotoredox catalyst, which is superior to all other methods. The range of substrates for visible light photoredox reactions has been expanded by this synthesis. The present strategy also provides many advantages of green chemistry, notably high atom economy, shortened reaction time, one-pot consolidated methodology, and high efficiency. Molecular docking studies carried out on the 4FGL protein associated with the oxidoreductase domain exhibited binding energy of − 5.9 kcal mol− 1 resulting it's application in the medicinal field.
- Koruzňjak D J, Grdisǎ M et al (2003) Novel Derivatives of Benzo[b]thieno[2,3-c]quinolones: Synthesis, Photochemical Synthesis, and Antitumor Evaluation. J Med Chem 46:4516−4524. https://doi.org/10.1021/jm0210966
- Acharya, A, Vijay K S, Saraiah,B, Ila H (2015) One-Pot Synthesis of Functionalized Benzo[b]thiophenes and Their Hetero-Fused Analogues via Intramolecular Copper-Catalyzed S-Arylation of In Situ Generated Enethiolates. J Org Chem 80:2884−2892. https://doi.org/10.1021/acs.joc.5b00032
- Acharya A, Kumar S V, Ila H (2015) Diversity-Oriented Synthesis of Substituted Benzo[b]thiophenes and Their Hetero-Fused Analogues through Palladium-Catalyzed Oxidative C-H Functionalization/Intramolecular Arylthiolation. Chem Eur J 21:17116-17125. https://doi.org/10.1002/chem.201501828
- Singh P P, Yadav A K, Ila H, Junjappa H (2009) Novel Route to 2,3-Substituted Benzo[b]thiophenes via Intramolecular Radical Cyclization. J Org Chem 74:5496-5501. https://doi.org/10.1021/jo900615p
- Keri R S, Chand K, Budagumpi S, Somappa S B, Patil S A, Nagaraja B M (2017) An overview of benzo[b]thiophene-based medicinal chemistry. Eur J Med Chem 138:1002-1033. https://doi.org/10.1016/j.ejmech.2017.07.038
- Palkowitz A D, Glasebrook A L, Thrasher K J, Hauser K L, Short L L., Phillips D L, Muehl B S, Sato M, Shetler P K, Cullinan G J, Pell T R, Bryant H U (1997) Discovery and Synthesis of [6-Hydroxy-3-[4-[2-(1-piperidinyl)ethoxy]phenoxy]- 2-(4-hydroxyphenyl)]benzo[b]thiophene: A Novel, Highly Potent, Selective Estrogen Receptor Modulator. J Med Chem 40:1407−1416. https://doi.org/10.1021/jm970167b
- Yang Y, Zhang T, Huang W, Shen Z (2014) Piperidine Nucleophilic Substitution Without Solvent: An Efficient Synthesis of Raloxifene. Synth Commun 44:3271−3276. https://doi.org/10.1080/00397911.2014.943348
- Dadiboyena, S (2012) Recent advances in the synthesis of raloxifene: A selective estrogen receptor modulator. Eur J Med Chem 51:17−34. https://doi.org/10.1016/j.ejmech.2012.02.021
- Hou C, He Q, Yang C (2014) Direct Synthesis of Diverse 2-Aminobenzo[b]thiophenes via Palladium-Catalyzed Carbon–Sulfur Bond Formation Using Na2S2O3 as the Sulfur Source. Org Lett 16:5040-5043. https://doi.org/10.1021/ol502381e
- Bolognese M, Krege J H, Utian W H, Feldman R, Broy S, Meats D L, Alam J, Lakshmanan M, Omizo M (2009) Effects of Arzoxifene on Bone Mineral Density and Endometrium in Postmenopausal Women with Normal or Low Bone Mass. J Clin Endocrinol Metab 94:2284−2289. https://doi.org/10.1210/jc.2008-2143
- Overk C R, Peng K W, Asghodom R T, Kastrati I, Lantvit D D, Qin Z, Frasor J, Bolton J L, Thatcher G R J (2007) Structure–Activity Relationships for a Family of Benzothiophene Selective Estrogen Receptor Modulators Including Raloxifene and Arzoxifene ChemMedChem 2:1520−1526. https://doi.org/10.1002/cmdc.200700104
- Abdelhamid R, Luo J, VandeVrede L, Kundu I, Michalsen B, Litosh V A, Schiefer I T, Gherezghiher T, Yao P, Qin Z, Thatcher G R J (2011) Benzothiophene Selective Estrogen Receptor Modulators Provide Neuroprotection by a Novel GPR30-Dependent Mechanism. ACS Chem Neurosci 2:256−268. https://doi.org/10.1021/cn100106a
- Hsiao C N, Kolasa T (1992) Synthesis of chiral zileuton, a potent and selective inhibitor of 5-lipoxygenase. Tetrahedron Lett 33:2629− 2632. https://doi.org/10.1016/S0040-4039(00)79043-5
- Basha A, Brooks D W J (1993) Synthesis of the 5-lipoxygenase inhibitor zileuton from thiophenol. Org Chem 58:1293− 1294. https://doi.org/10.1021/jo00057a056
- Bando Y, Tohyama H, Aoki K, Kanehara H, Hisada A, Okafuji K, Toya D (2016) Ipragliflozin lowers small, dense low-density lipoprotein cholesterol levels in Japanese patients with type 2 diabetes mellitus. J Clin Transl Endocrinol 6:1−7. https://doi.org/10.1016/j.jcte.2016.06.001
- Lu C H, Min K W, Chuang L M, Kokubo S, Yoshida S, Cha B S (2016) Efficacy, safety, and tolerability of ipragliflozin in Asian patients with type 2 diabetes mellitus and inadequate glycemic control with metformin: Results of a phase 3 randomized, placebo-controlled, double-blind, multicenter trial. J Diabetes Investig 7:366−373. https://doi.org/10.1111/jdi.12422
- Poole R M, Dungo R T (2014) Ipragliflozin: First Global Approval. Drugs 74:611−617. https://doi.org/10.1007/s40265-014-0204-x
- Croxtall J D, Plosker G L (2009) A Review of Its Use in the Management of Superficial Mycoses in Dermatology and Gynaecology. Drugs 69:339−359. https://doi.org/10.2165/00003495-200969030-00009
- Treǵuier B, Lawson M, Bernadat G, Bignon J, Dubois J, Brion J D, Alami M, Hamze A (2014) Synthesis of a 3-(α-Styryl)benzo[b]-thiophene Library via Bromocyclization of Alkynes and Palladium-Catalyzed Tosylhydrazones Cross-Couplings: Evaluation as Antitubulin Agents. ACS Comb Sci 16:702−710. https://doi.org/10.1021/co500115b
- Romagnoli R, Baraldi P G, Lopez-Cara C, Preti D, Aghazadeh Tabrizi M, Balzarini J, Bassetto M, Brancale A, Fu X H, Gao Y, Li J, Zhang S Z, Hamel E, Bortolozzi R, Basso G, Viola G (2013) Concise Synthesis and Biological Evaluation of 2-Aroyl-5-Amino Benzo[b]thiophene Derivatives As a Novel Class of Potent Antimitotic Agents. J Med Chem 56:9296−9309. https://doi.org/10.1021/jm4013938
- Lim C J, Woo S E, Ko S I, Lee B H, Oh K S, Yi K Y (2016) Benzo[b]thiophene-2-carboxamide derivatives as potent urotensin-II receptor antagonists. Bioorg Med Chem Lett 26:4684−4686. https://doi.org/10.1016/j.bmcl.2016.08.049
- Hur W, Rosen H, Gray N S (2017) A benzo[b]thiophene-based selective type 4 S1P receptor agonist. Bioorg Med Chem Lett 27:1−5. https://doi.org/10.1016/j.bmcl.2016.11.050
- Mahajan P S, Nikam M D, Nawale L U, Khedkar V M, Sarkar D, Gill C H (2016) Synthesis and Antitubercular Activity of New Benzo[b]thiophenes. ACS Med Chem Lett 7:751−756. https://doi.org/10.1021/acsmedchemlett.6b00077
- Fakhr I M I, Radwan M A A, El-Batran S, Abd El-Salam O M E, El-Shenawy S M (2009) Synthesis and pharmacological evaluation of 2-substituted benzo[b]thiophenes as anti-inflammatory and analgesic agents. Eur J Med Chem 44:1718−1725. https://doi.org/10.1016/j.ejmech.2008.02.034
- Iino H, Usui T, Hanna Ji (2015) Liquid crystals for organic thin-film transistors. Nat Commun 6:6828. https://doi.org/10.1038/ncomms7828
- Iino H, Hanna Ji (2017) Liquid crystalline organic semiconductors for organic transistor applications. Polym J 49:23−30. https://doi.org/10.1038/pj.2016.101
- Du Z, Chen W, Qiu M, Chen Y, Wang N, Wang T, Sun M, Yu D, Yang R (2015) Utilizing alkoxyphenyl substituents for side-chain engineering of efficient benzo[1,2-b:4,5-b′]dithiophene-based small molecule organic solar cells. Phys Chem Chem Phys 17:17391−17398. https://doi.org/10.1039/C5CP02632F
- Sista P, Biewer M C, Stefan M C (2012) Benzo[1,2-b:4,5-b′]dithiophene Building Block for the Synthesis of Semiconducting Polymers. Macromol Rapid Commun 33:9−20. https://doi.org/10.1002/marc.201100671
- Capodilupo A L, Fabiano E, De Marco L, Ciccarella G, Gigli G, Martinelli C, Cardone A (2016) [1]Benzothieno[3,2-b]benzothiophene-Based Organic Dyes for Dye-Sensitized Solar Cells. J Org Chem 81:3235−3245. https://doi.org/10.1021/acs.joc.6b00192
- Gabriele B, Mancuso R, Lupinacci E, Veltri L, Salerno G, Carfagna C (2011) J Org Chem 76:8277-8286. https://doi.org/10.1021/jo201471k
- Yugandar S, Konda S, Ila H (2017) Synthesis of Substituted Benzo[b]thiophenes via Sequential One-Pot, Copper-Catalyzed Intermolecular C–S Bond Formation and Palladium-Catalyzed Intramolecular Arene–Alkene Coupling of Bis(het)aryl/alkyl-1,3-monothiodiketones and o-Bromoiodoarenes. Org Lett 19:1512-1515. https://doi.org/10.1021/acs.orglett.7b00273
- Gao F, Wang J T, Liu L L, Ma N, Yang C, Gao Y, Xia W (2017) Synthesis of carbonylated heteroaromatic compounds via visible-light-driven intramolecular decarboxylative cyclization of o-alkynylated carboxylic acids. Chem Commun 53:8533-8536. https://doi.org/10.1039/C7CC04813K
- Liu W, Hu Y Q, Hong X Y, Li G X, Huang X B, Gao W X, Liu M C, Xia Y, Zhou Y B, Wu H Y (2018) Direct synthesis of 3-acylbenzothiophenes via the radical cyclization of 2-alkynylthioanisoles with α-oxocarboxylic acids. Chem Commun 54:14148-14151. https://doi.org/10.1039/C8CC07735E
- Masuya Y, Tobisu M, Chatani N (2016) Palladium-Catalyzed Synthesis of 2,3-Disubstituted Benzothiophenes via the Annulation of Aryl Sulfides with Alkynes. Org Lett 18:4312-4315. https://doi.org/10.1021/acs.orglett.6b02055
- Kwak S H, Lim S J, Yoo H J, Ha J E, Gong Y D (2016) Intramolecular Mizoroki–Heck Reaction of 2-Thiosubstituted Acrylates for the Synthesis of 3-Substituted Benzo[b]thiophene-2-carboxylates. Synthesis 48:4131-4142. DOI: 10.1055/s-0035-1562613
- Garg P, Singh A (2018) Unmasking Dipole Character of Acyl Ketene Dithioacetals via a Cascade Reaction with Arynes: Synthesis of Benzo[b]thiophenes. Org Lett 20:1320−1323. https://doi.org/10.1021/acs.orglett.8b00053
- Kumar Y, Ila H (2021) Synthesis of Substituted Benzo[b]thiophenes via Base-Promoted Domino Condensation−Intramolecular C−S Bond Formation. Org Lett 23:1698−1702. https://dx.doi.org/10.1021/acs.orglett.1c00085
- Alikhani Z, Albertson A. G, Walter C A, Masih P J, Kesharwani T (2022) Synthesis of Benzo[b]thiophenes via Electrophilic Sulfur Mediated Cyclization of Alkynylthioanisoles. J Org Chem 87:6312–6320. https://doi.org/10.1021/acs.joc.1c02606
- Srinivas B, Shakeena K, Kota D L, Abhinav V, Eswar P, Sravani R G, Kumar A S P, Indukuri K, Dhanaraju K A, Kumar M M K, Alla S K (2023) Iron(III)-Catalyzed Regioselective Synthesis of Electron-Rich Benzothiazoles from Aryl Isothiocyanates via C–H Functionalization. J Org Chem 88:4458–4471. https://doi.org/10.1021/acs.joc.2c03078
- Hari D P, König B (2014) Synthetic applications of eosin Y in photoredox catalysis. Chem Commun 50:6688-6699. https://doi.org/10.1039/C4CC00751D
- Heitz D R, Rizwan K, Molander G A (2016) Visible-Light-Mediated Alkenylation, Allylation, and Cyanation of Potassium Alkyltrifluoroborates with Organic Photoredox Catalysts. J Org Chem 81:7308-7313. https://doi.org/10.1021/acs.joc.6b01207
- Emmanuel M A, Bender S G, Bilodeau C, Carceller J M, DeHovitz J S, Fu H, Liu Y, Nicholls B T, Ouyang Y, Page C G, Qiao T, Raps F C, Sorigué D R, Sun S Z, Turek-Herman J, Ye Y, Rivas-Souchet A, Cao J, Hyster T K (2023) Photobiocatalytic Strategies for Organic Synthesis. Chem Rev https://doi.org/10.1021/acs.chemrev.2c00767
- Candish L, Collins K D, Cook G C, Douglas J J, Jolit A G A, Keess S (2022) Photocatalysis in the Life Science Industry. Chem Rev 122:2907-2980. https://doi.org/10.1021/acs.chemrev.1c00416
- Sahoo A K, Rakshit A, Dahiya A, Pan A, Patel B K (2022) Visible-Light-Mediated Synthesis of Thio-Functionalized Pyrroles. Organic Letters 24:1918-1923. https://doi.org/10.1021/acs.orglett.2c00283
- Ghosh A, Pyne P, Ghosh S, Ghosh D, Majumder S, Hajra A (2022) Visible-light-induced metal-free coupling of C(sp3)–H sources with heteroarenes. Green Chem 24:3056-3080. https://doi.org/10.1039/D1GC04384F
- Das S, Azim A, Hota S K, Panda S P, Murarka S, Sarkar S D (2021) An organophotoredox-catalyzed redox-neutral cascade involving N-(acyloxy)phthalimides and allenamides: synthesis of indoles. Chem Commun 57:13130-13133. https://doi.org/10.1039/D1CC05397C
- Wang H, Wu Q, Zhang J D, Li Y H, Li H X (2021) Photocatalyst- and Transition-Metal-Free Visible-Light-Promoted Intramolecular C(sp2)–S Formation. Organic Letters 23:2078-2083. https://doi.org/10.1021/acs.orglett.1c00235
- Yu W Q, Xie J, Chen Z, Xiong B Q, Liu Y, Tang K W (2021) Visible-Light-Induced Transition-Metal-Free Nitrogen-Centered Radical Strategy for the Synthesis of 2-Acylated 9H-Pyrrolo[1,2-a]indoles. J Org Chem 86:13720–13733. https://doi.org/10.1021/acs.joc.1c01834
- Srivastava V, Singh P K, Singh P P (2015) Eosin Y Catalysed Visible light promoted aerobic oxidative cyclization of 2-Aminobenzothiazole. Croat Chem Acta 88:227–233. DOI: 10.5562/cca2632
- Srivastava V, Singh P P (2017) Eosin Y catalyzed photo- redox synthesis: a review. RSC Adv 7:31377–31392. https://doi.org/10.1039/C7RA05444K
- Srivastava V, Singh P K (2018) Kanaujia S, Singh P P, Photoredox catalysed synthesis of amino alcohol. New J Chem 42:688–691. https://doi.org/10.1039/C7NJ03068A
- Singh P K, Singh P P, Srivastava V (2018) Facile aerobic photo oxidative synthesis of sulfinic esters. Croat Chem Acta 91:383–387. https://doi.org/10.5562/cca3401
- Srivastava V, Singh P K, Singh P P (2019) Visible light pho- toredox catalyzed amidation of carboxylic acids with amines. Tetrahedron Lett 60:40–43. https://doi.org/10.1016/j.tetlet.2018.11.050
- Srivastava V, Singh P K, Singh P P (2019) Photocatalysed eosin Y mediated C(sp3)-H alkylation of amine substrates via direct HAT. Tetrahedron Lett 60:1333–1336. https://doi.org/10.1016/j.tetlet.2019.04.016
- Srivastava V, Singh P K, Singh P P (2019) Eosin Y catalysed visible-light mediated aerobic oxidation of tertiary amines. Tetrahedron Lett 60:151041. https://doi.org/10.1016/j.tetlet.2019.151041
- Srivastava V, Singh P K, Srivastava A, Singh P P (2020) Recent application of visible light induced radicals in C-S bond formation. RSC Adv 10:20046–20056. https://doi.org/10.1039/D0RA03086D
- Srivastava V, Singh P K, Singh P P (2020) Visible light promoted synthesis of disubstituted 1,2,3-thiadiazoles. Rev Roum Chim 65:221–226. https://doi.org/10.33224/rrch.2020.65.3.01
- Srivastava A, Singh P K, Ali A et al (2020) Recent applications of rose bengal catalysis in N-heterocycles: a short review. RSC Adv 10:39495–39508. https://doi.org/10.1039/D0RA07400D
- Srivastava V, Singh P K, Srivastava A, Singh P P (2021) Synthetic applications of flavin photocatalysis: a review. RSC Adv 11:14251–14259. https://doi.org/10.1039/D1RA00925G
- Srivastava V, Singh PP (2021) Recent advances of 4DPAIPN in photocatalytic transformations. Org Biomol Chem 19:313–321. https://doi.org/10.1039/D0OB01884H
- Singh PP, Singh PK, Beg MZ et al (2021) Recent applications of photoredox catalysis in O-heterocycles: a short review. Synth Commun 51:3033–3058. https://doi.org/10.1080/00397911.2021.1968907
- Srivastava V, Singh P K, Srivastava A et al (2021) Recent advances of dicyanopyrazine (DPZ) in photoredox catalysis. Photochem 1:237–246. https://doi.org/10.3390/photochem1020014
- Srivastava V, Singh P K, Tivari S, Singh P P (2022) Visible light photocatalysis in the synthesis of pharmaceutically relevant heterocyclic scaffolds. Org Chem Front 9:1485–1507. https://doi.org/10.1039/D1QO01602D
- Srivastava V, Singh P K, Singh P P (2022) Recent advances of visible-light photocatalysis in the functionalization of organic compounds. J Photochem Photobiol C 50:100488. https://doi.org/10.1016/j.jphotochemrev.2022.100488
- Singh P P, Srivastava V (2022) Recent advances in visible-light graphitic carbon nitride (g-C3N4) photocatalysts for chemical transformations. RSC Adv 12:18245–18265. https://doi.org/10.1039/D2RA01797K
- Singh P P, Sinha S, Pandey G, Srivastava V (2022) Molybdenum disulfide (MoS2) based photoredox catalysis in chemical transformations. RSC Adv 12:29826- 29839. https://doi.org/10.1039/D2RA05695J
- Tivari S, Singh P K, Singh P P, Srivastava V (2022) Visible light-induced photoredox catalyzed C–N coupling of amides with alcohols. RSC Adv 12:35221. https://doi.org/10.1039/D2RA07065K
- Mishra M, Srivastava V, Singh P K, Singh P P (2022) Visible-light Driven Eosin Y Catalyzed C(sp2)-H Functionalization/C–O Bond Formation for Synthesis of Benzoxazoles. Croat Chem Acta 95:25–30. DOI: 10.5562/cca3927
- Singh P P, Singh P K, Srivastava V (2023) Visible light metal- laphotoredox catalysis in the late-stage functionalization of pharmaceutically potent compounds. Org Chem Front 10:216-236. https://doi.org/10.1039/D2QO01582J
- Singh S P, Srivastava V, Singh P K, Singh P P (2023) Visible-light induced eosin Y catalysed C(sp2)-H alkylation of carbonyl substrates via direct HAT. Tetrahedron 132:133245. https://doi.org/10.1016/j.tet.2023.133245
- Beg M Z, Singh P K, Singh P P, Srivastava M, Srivastava V (2023) Metal‐free visible light mediated direct C–H amination of benzoxazole with secondary amines. Molecular Diversity https://doi.org/10.1007/s11030-022-10595-2
- Singh P P, Singh J, Srivastava V (2023) Visible-light acridinium-based organophotoredox catalysis in late-stage synthetic applications. RSC Adv 13:10958–10986. https://doi.org/10.1039/D3RA01364B
- Srivastava V, Tivari S, Singh P K, Singh P P (2023) Photocatalysed Synthesis and Structure Activity Evaluation of Cyclohexyloxyphenethylpyridinones as Potent HIV‐1 Inhibitor. Catalysis Letters https://doi.org/10.1007/s10562-023-04345-8
- Srivastava V, Sinha S, Kumar D, Singh P P (2023) Recent chemical transformation involving gold based photocatalysis. Tetrahedron Green Chem
- Lambert C R, Kochevar I E (1996) Does Rose Bengal Triplet Generate Superoxide Anion? J Am Chem Soc 118:3297–3298. https://doi.org/10.1021/ja9600800
- Encinas M V, Rufs A M, Bertolotti S G, Previtali C M (2009) Xanthene dyes/amine as photoinitiators of radical polymerization: A comparative and photochemical study in aqueous medium. Polymer 50:2762-2767. https://doi.org/10.1016/j.polymer.2009.04.024
- Lizarides T, McCormick T, Du P, Luo G, Lindley B, Eisenberg R (2009) Making Hydrogen from Water Using a Homogeneous System Without Noble Metals. J Am Chem Soc 131:9192-9194. https://doi.org/10.1021/ja903044n
- Lee S H, Nam D H, Park C B (2009) Screening Xanthene Dyes for Visible Light-Driven Nicotinamide Adenine Dinucleotide Regeneration and Photoenzymatic Synthesis. Adv Synth Catal 351:2589-2594. https://doi.org/10.1002/adsc.200900547
- Xiao T, Dong X, Tang Y, Zhou L (2012) Phenanthrene Synthesis by Eosin Y-Catalyzed, Visible Light- Induced [4+2] Benzannulation of Biaryldiazonium Salts with Alkynes. Adv Synth Catal 354:3195-3199. https://doi.org/10.1002/adsc.201200569
- Fekarurhobo G K, Angaye S S, F. G. Obomann F G (2013) Methylene Blue-Sensitized Photoxidation of Olive Oil. Journal of Emerging Trends in Engineering and Applied Sciences (JETEAS) 4(3):394-401.
- Udoikono A D, Louis H, Eno E A, Agwamba E C, Unimuke T O, Igbalagh A T, Edet H O, Odey J O, Adeyinka A S (2022) Reactive azo compounds as a potential chemotherapy drugs in the treatment of malignant glioblastoma (GBM): Experimental and theoretical studies. J. Photochem. Photobiol. 10:100116. https://doi.org/10.1016/j.jpap.2022.100116
- Siddiqui A, Khan J, Hamadou M F, Sabri W et al (2021) Molecular docking and dynamics simulation revealed ivermectin as potential drug against schistosoma-associated bladder cancer targeting protein signaling: Computational drug repositioning approach. Medicina 57:1058, https://doi.org/10.3390/medicina57101058
- Mujwar S (2021) Computational repurposing of tamibarotene against triple mutant variant of SARS-CoV-2. Comput Biol Med 136:104748. https://doi.org/10.1016/j.compbiomed.2021.104748
- Pandey A K, Shukla D V, Mishra V N, Singh V, Yadav O P, Dwivedi A (2022) Molecular docking, experimental FT-IR spectra, UV–Vis spectra, vibrational analysis, electronic properties, Fukui function analysis of a potential bioactive agent – Proflavine. J Indian Chem. Soc 99:100396. https://doi.org/10.1016/j.jics.2022.100396
- Mujwar S, Kumar V (2020) Computational drug repurposing approach to identify potential fatty acid-binding protein-4 inhibitors to develop novel antiobesity therapy. Assay and Drug Development Technologies 18:318–3271. https://doi.org/10.1089/adt.2020.976
- Douche D, Sert Y, Brandán S A, Kawther A A, Bilmez B, Dege A, Louzi A E, Bougrin K, Karrouchi K, Himmi B (2021) 5-((1H-imidazol-1-yl) methyl) quinolin-8-ol as potential antiviral SARS-CoV-2 candidate: Synthesis, crystal structure, Hirshfeld surface analysis, DFT and molecular docking studies. J Mol Struct 1232:130005. https://doi.org/10.1016/j.molstruc.2021.130005
- Leung K K K, Shilton B H (2013) Chloroquine Binding Reveals Flavin Redox Switch Function of Quinone Reductase 2. J Biol Chem 288:11242–11251. https://doi.org/10.1074/jbc.M113.457002
- Li H, Wang H, Kang S, Silverman R B, Poulos T L (2016) Electrostatic Control of Isoform Selective Inhibitor Binding in Nitric Oxide Synthase. Biochemistry 55:3702–3707. https://doi.org/10.1021/acs.biochem.6b00261
- Lipinski C A, Lombardo F, Dominy B W, Feeney P J (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25. https://doi.org/10.1016/S0169-409X(96)00423-1
- Daina A, Michielin O, Zoete V (2014) iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J Chem Inf Model 54:3284–3301. https://doi.org/10.1021/ci500467k
Table 2 is available in the Supplementary Files section.
Schemes 1 and 2 are available in the Supplementary Files section.
No competing interests reported.
- Table2.docx
- SIFile.pdf
- GA.png
- scheme1.png
Scheme 1. Synthesis of benzo[b]thiophenes.
- scheme2.png
Scheme 2. Plausible mechanism for visible-light mediated photocatalysed synthesis of benzo[b]thiophenes.
{kind=link}
{kind=link}
{kind=link}