2.1. Synthesis and characterization of poly(ethylene glycol)-b-poly(epsilon-caprolactone) block copolymer
A PEG-b-PCL triblock copolymer was synthesized via a ring opening polymerization (ROP) reaction of epsilon-caprolactone (CL, Sigma-Aldrich, St. Louis, MO, USA) initiated by the terminal hydroxyl group of the monofunctional methoxy PEG (mPEG4000, molecular weight – MW – 4000 g/mol, Tokyo Chemical Industry Co., Ltd, Tokyo, Japan) in the presence of tin(II) 2-ethylhexanoate (Sn(Oct)2, Sigma-Aldrich) as catalyst. Both CL and Sn(Oct)2 were dried prior to use with molecular sieves 3A (Alfa Aesar, Ward Hill, MA, USA) pre-activated at 150°C (4 h) at least 24 h before use. Feed weights of the reactants were calculated based on the desired MW of the PCL block (~ 30,000 g/mol). For this, mPEG (0.5 g) was weighed, poured into a round-bottom flask with a magnetic stirring bar, melted at 100°C, dried under vacuum for 2 h, allowed to cool at room temperature (RT) and the flask was sealed with a septum under dry nitrogen environment conditions, which were maintained throughout the reaction. Next, CL (3.75 g) was added to the liquid by injection through the septum, the reaction mixture heated to 145°C, Sn(Oct)2 (1:200 molar ratio to CL) added to the reaction mixture and the reaction allowed to proceed at 145°C for 2.5 h. The reaction mixture was cooled to RT, dissolved in dichloromethane (20 mL, Sigma-Aldrich) and precipitated in an excess of diethyl ether (500 mL). The precipitated PEG-b-PCL copolymer was filtered in a Büchner funnel using filter paper (Whatman™ 1, Sigma Aldrich) to remove remaining unreacted reagents, washed several times with diethyl ether, vacuum-dried at RT (Vacuum Oven Lab-Line Instruments Inc., Dubuque, IL, USA) until constant weight and stored at -24ºC until use.
The weight-average molecular weight (Mw), the number-average molecular weight (Mn) and the dispersity (Ð, Mw/Mn) of the PEG-b-PCL copolymer were measured by gel permeation chromatography (GPC) in a Viscotek 270max GPC system (Malvern Instruments, Malvern, UK) equipped with refractive index, viscometer, and light scattering detectors. The copolymer was dissolved in tetrahydrofuran (THF, 5 mg/mL) for 24 h and a THF solution containing 200 ppm of butylated hydroxytoluene was used as the mobile phase. The injection volume was 100 µL and the chromatographic separation was performed using three LT4000L - Mixed bed (300 x 8.0 mm) columns at a flow rate of 1.0 mL/min and a temperature of 35°C. The instrument calibration was done with 105,000 and 245,000 g/mol polystyrene standards (Malvern Instruments).
The proton-nuclear magnetic resonance (1H-NMR) spectrum of the copolymer was recorded in a 1% w/v solution of deuterated acetone (acetone-d6, Cambridge Isotope Laboratories, Inc., Tewksbury, MA, USA). Chemical shifts are reported in ppm using the signal of acetone at 2.05 ppm as internal standard.
PEG-b-PCL was also characterized by attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR) and spectra were recorded in a Tensor 27 spectrometer (Bruker Optics, Inc., Rheinstetten, Germany) from 4000 to 400 cm− 1 (32–64 scans with a resolution of 4 cm− 1).
2.2. Synthesis and characterization of poly(ethylene glycol)-b-poly(epsilon-caprolactone) nanoparticles
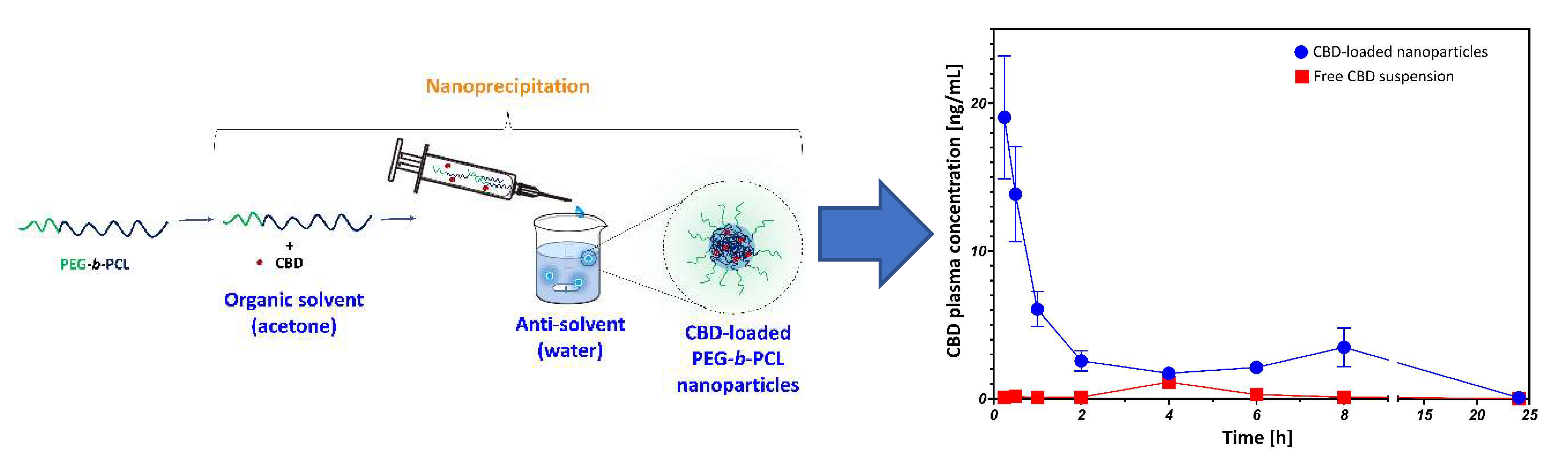
PEG-b-PCL nanoparticles were synthesized using a nanoprecipitation method with homogenization. Briefly, PEG-b-PCL (50 mg) was dissolved in acetone (10 mL, Bio-Lab Ltd., Jerusalem, Israel), a beaker with deionized water (50 mL, the anti-solvent) was put in a water bath with controlled temperature (23 ºC, Hei-Tec Magnetic Stirrer, 600 rpm, Heidolph Instruments, Schwabach, Germany) and homogenized at 12,100 rpm with a homogenizer (POLYTRON® PT 2500 E, Kinematica AG, Luzern, Switzerland). Then, the acetone solution of the copolymer was added at once, homogenized for 16 min and kept under constant magnetic stirring (600 rpm) for 6 h to allow complete acetone evaporation. The nanoparticle suspension was kept at 4ºC until use. To produce CBD-loaded nanoparticles, CBD (6 mg) was added to the copolymer solution in acetone (50 mg in 10 mL), allowed to dissolve overnight protected from light and the process conducted as described above.
To quantify the CBD encapsulation efficiency (%EE) and loading (%DL) in the nanoparticles, we used analytical reversed phase ultra-high performance liquid chromatography-ultraviolet detector (HPLC-UV, UltiMate 3000, Thermo Scientific UHPLC, Bremen, Germany) equipped with a photodiode array (PDA) detector with a Kinetex C18 core-shell column (2.6 µm, 150 mm×2.1 mm i.d.) and a guard column (0.5 µm depth filter×0.1 mm, Phenomenex, Torrance, CA, USA) and an A/B isocratic method (solvent A: 0.1% acetic acid in water, solvent B: 0.1% acetic acid in acetonitrile). The isocratic program conditions were: 15% A and 85% B for 5 min. A flow rate of 0.25 mL/min was used, the column temperature was held at 30°C and the injection volume was 1–4 µL. Liquid chromatography/mass spectrometry (LC/MS) grade acetonitrile, water for the mobile phase and HPLC grade EtOH for sample preparation were obtained from Mercury Scientific and Industrial Products Ltd. (Rosh Ha’ayin, Israel). LC/MS grade acetic acid was purchased from Sigma-Aldrich (Rehovot, Israel). The CBD content quantified by HPLC-UV was used to calculate %EE according to Eq. 1 and the %DL according to Eq. 2
% EE =\(\frac{Total CBD in the nanoparticles}{Total CBD weight used}x 100\) (1)
% DL \(= \frac{Total encapsulated CBD}{Copolymer weight + CBD weight}x 100\)(2)
Each nanoparticle suspension was injected to the HPLC-UV in duplicates and results expressed as mean ± S.D of quintuplicates (n = 5).
The hydrodynamic diameter (Dh) and polydispersity index (PDI) of CBD-free and CBD-loaded PEG-b-PCL nanoparticles (0.1% w/v of nanoparticles in suspensions) were measured at 25 and 37°C by dynamic light scattering (DLS, Zetasizer Nanoseries ZS90, Malvern Instruments) at a scattering angle of 173°. Results are expressed as mean ± S.D. of at least three measurements. DLS data were analyzed using CONTIN algorithms (Malvern Instruments). Zeta-potential (Z-potential) measurements were carried out by using laser Doppler micro-electrophoresis in the same instrument.
The morphology of CBD-loaded nanoparticles was visualized by high resolution-scanning electron microscopy (HR-SEM, 0.02-30 kV Zeiss Ultra-Plus FEG-SEM, Carl Zeiss NTS GmbH, Oberkochen, Germany). Samples were prepared by spraying 20 µL of a sample on a silicon wafer (CZ polished silicon wafers < 100 > oriented, highly doped N/Arsenic, SEH Europe Ltd., Livingston, UK) by using nitrogen gas flow to cast small droplets over the wafer, and by doing so, ensuring a thin layer of particles distributed on it. The suspension was used immediately after nanoprecipitation and did not undergo freeze-drying. This process was repeated several times, and the sample was allowed to dry for at least 24 h at RT and atmospheric pressure. The acceleration voltage was 1.5 kV, and the images were acquired using in-lens secondary electrons at 2–3 mm working distance. The size of 25 nanoparticles was measured using ImageJ 1.8 software (National Institutes of Health, Bethesda, MD, USA), and expressed as mean ± S.D.
Imaging was also performed by high resolution-cryogenic-transmission electron microscopy (HR-cryo-TEM) in a FEI T12 G2 electron microscope (FEI, Hillsboro, Oregon, US) operated at 120 kV. Images were recorded digitally by a Gatan US 1000 high-resolution CCD camera (Tecnai T12 G2, FEI), using the DigitalMicrograph® software. A drop (3 µL) of each sample was placed on a carbon-coated perforated polymer film, supported on a 200 mesh TEM grid (Ted Pella, Inc., Redding, CA, USA), and mounted on a tweezer. The drop was turned into a thin film (preferably less than 300 nm) by blotting away excess solution with a filter paper-covered metal strip. The grid was then plunged quickly into liquid ethane at its freezing point (183°C). Prior to the specimen preparation, grids were plasma etched in a PELCO EasiGlow glow- discharger (Ted Pella, Inc.) to increase their hydrophilicity. The size of 25 nanoparticles was measured using ImageJ 1.8 software, and expressed as mean ± S.D.
2.3. CBD release in vitro
Since the CBD-loaded nanoparticles are envisioned for oral administration, the CBD release in vitro was studied under gastric-like (pH = 1.2, 2 h, n = 3) and intestine-like conditions (pH 6.8, 24 h, n = 3).
For gastric-like conditions media of 0.1N HCl (37%, 0.1M pH = 1.2, Merck Chemicals GmbH) containing 0.1% w/v TWEEN® 80 (Merck Chemicals GmbH). The solution was prepared as follows: 4 mL of HCl was added to 125 mL of distilled water and the final volume adjusted to 500 mL with distilled water; intestine-like conditions were obtained by using 0.1M PBS (pH = 6.8) containing 0.1% w/v TWEEN® 80 for 0–24 h. The PBS was prepared as follows: 87 g of potassium phosphate dibasic (K2HPO4, Spectrum Chemical MFG Corp., Gardena, CA, USA) and 68 g of potassium phosphate mono basic (KH2PO4, Merck Chemicals GmbH) were dissolved separately in 0.5 L of distilled water and mixed until complete dissolution. Then, 50.3 mL of KH2PO4 and 49.7 mL of K2HPO4 were added to a 1 L measuring flask and distilled water was added to complete 1 L.
For both the gastric- and intestine-like release tests, 1.35 mL of nanoparticle suspension (CBD concentration of 0.12 mg/mL) was added to 250 mL of the release medium and the suspension were shaken (150 rpm) in an Excella E25 shaker incubator (New Brunswick Scientific, Edison, NJ, USA) at 37 ºC. At different timepoints, aliquots were taken (1 mL, was done in duplicates) and pre-heated fresh release medium was added to maintain a constant volume. Aliquots were ultracentrifuged at 180,000 G, at 4ºC, for 30 min (Sorvall Discovery M120 SE Micro-Ultracentrifuge, Thermo Fisher Scientific). Then, two aliquots of 300 µL were taken at each timepoint, frozen and dried by SpeedVac for 13 h, followed by CBD extraction from each with 300 µL of EtOH. Each CBD solution in EtOH obtained from the extraction of one release aliquot was injected into the HPLC-UV (as detailed above) in duplicates for CBD quantification, and the results expressed as mean ± S.D.
The CBD release rate profile data were fitted using Higuchi, first-order, and Hixson-Crowell models; our results did not fit them. The Korsmeyer-Peppas model is very popular, though only release data up to 60% can be used, which was not possible in this work due to the faster CBD release. Thus, we modeled our data to a hyperbolic tangent (tanh) function model proposed by Eltayeb et al. [49] that is based on the diffusive release model by Peppas and collaborators, but approximates the release to 100%, as expressed by Eq. 3
$${Q}_{t}={Q}_{\infty }\text{tanh}\left(\alpha {t}^{0.5}\right)\left(3\right)$$
Where Q∞ is the total fraction of CBD released from the nanoparticles, Qt is the fraction released in time t and α is a constant related to the particle size and diffusion constant. The model fitting was done with Python.
2.4. Cryo/lyoprotection of the CBD-loaded nanoparticles
To stabilize the CBD-loaded nanoparticles during freeze-drying and enable redispersion towards in vivo studies, 2-hydroxy-propyl-beta-cyclodextrin (Hpβ-CD, Glentham Life Sciences, Corsham, UK) was tested as a cryo/lyoprotectant. For this, Hpβ-CD was added to the CBD-loaded nanosuspension (DL% = 11%, CBD concentration of 0.012% w/v and nanoparticle concentration of 0.1% w/v) and their ability to preserve the nanoparticles was assessed in a three-step experiment. After each step, the Dh (measured by intensity and number) and the PDI were measured by DLS at 25°C. The first step comprised the addition of the cryo/lyoprotectant at concentrations of 1–15% w/v. In the second, the cryo/lyoprotection efficiency was assessed in a freezing/thawing cycle at -196 and 24°C, respectively. For this, the cryo/lyo-protectant was added to the CBD-loaded nanoparticle suspensions, the system frozen in liquid nitrogen, thawed at RT and the Dh and PDI of the nanoparticles before and after the freeze-thawing cycle compared. The cryo/lyoprotectant concentration that preserved the nanoparticle properties was used for the third step that was the freeze-drying of the suspension for 72 h (Labconco Free Zone 4.5 plus L Benchtop Freeze Dry System, Labconco Corp., Kansas City, MO, USA), and its resuspension. Based on that assay, Hpβ-CD concentrations of 2.5%, 4.5% and 6% w/v were chosen to cryo/lyoprotect and concentrate the CBD-loaded nanoparticle suspension upon freeze-drying and redispersion by 6.5 and 12.5-fold for the PK study. nanoparticles were characterized with DLS at 25°C.
2.5. Comparative oral CBD pharmacokinetics in rat
A pharmacokinetic (PK) study was conducted at “Science in Action” (Ness Ziona, Israel) and following the ethical guidelines of animal experimentation. Male rats (Sprague Dawley, ~ 200 g, 7–8 weeks old) were allowed to acclimatize for one week, then fasted for 4 h with free access to water, and randomly divided into two groups (n = 5). The control group was administered by gavage 2 mL of a pure CBD suspension prepared as followed: 50 mL water was added to 72.5 mg CBD (i.e., 2.9 mg CBD per gavage) and 9.2 mg of Cremophor EL® (Merck Chemicals GmbH), vortexed and the suspension was stirred for 30 min with a magnetic stirrer (300 rpm). The treatment group was administered 2 mL of CBD-loaded nanoparticle suspensions prepared as described before (section 2.2) with CBD concentration of 0.12 mg/mL. Then, the nanosuspension (24 mL) was added to 1.1 g of Hpβ-CD, vortexed, and freeze-dried (Labconco Free Zone 4.5 plus L Benchtop Freeze Dry System) for 72 h. Prior to the gavage administration, the dry powder of CBD-loaded nanoparticles and cryo/lyoprotectant containing 2.9 mg CBD was re-suspended in water by vortex and magnetic stirring (300 rpm, 30 min). Both rat groups were administered a CBD dose of 14.5 mg/kg. Blood samples (100 µL) were taken from the cheek vein at 0.25, 0.5, 1, 2, 4, 6, 8 and 24 h post-administration. Then, rats were euthanized by CO2 inhalation.
The quantification of CBD, 7-hydroxy CBD (7-OH-CBD) and 7-carboxy-CBD (7-COOH-CBD) was done in collaboration with Cannasoul Analytic Ltd. (Caesarea, Israel) by using a previously validated sample preparation and HPLC-MS methods [50].
Briefly, blood samples were centrifuged for 30 min at 4°C, plasma was extracted by a vigorous vortex with a solution consisting of methanol:acetonitrile:acetic acid at a 50:50:0.1 volume ratio and spiked with CBD, 7-OH-CBD and 7-COOH-CBD deuterated internal standards. All samples were centrifuged (14,000 rpm) for 20 min at 4°C. Volumes of 0.7 mL were collected from plasma supernatants, diluted with 3 mL 0.1% v/v acetic acid in water, and loaded onto C8 solid phase extraction cartridges and loaded onto pre-conditioned Agela Cleanert C8 solid phase extraction cartridges (SPE, 500 mg of sorbent, 50 µm particle size, Agela Technologies, CA, USA) using 5 mL of 0.1% v/v acetic acid in methanol followed by 3 mL of water with 0.1% v/v acetic acid. CBD was eluted from the SPE columns with 2 mL 0.1% v/v acetic acid in methanol, evaporated to dryness by SpeedVac, reconstituted in 100 µL ethanol and filtered through a 0.22 µm PTFE syringe filter (Silicol Scientific Equipment Ltd., Or Yehuda, Israel).
HPLC-MS analyses were performed using a Thermo Scientific ultra HPLC system coupled with a Q Exactive™ Focus Hybrid Quadrupole-Orbitrap MS (Thermo Scientific). The chromatographic separation was achieved using a Halo C18 Fused Core column (2.7 µm, 150 mm × 2.1 mm i.d.) with a guard column (2.7 µm, 5 mm × 2.1 mm i.d) (Advanced Materials Technology, Wilmington, DE, USA) and a ternary A/B/C multistep gradient (solvent A: 0.1% acetic acid in water, solvent B: 0.1% acetic acid in acetonitrile, and solvent C: methanol). The multistep gradient program was set as follows: Initial conditions were 50% B raised to 67% B until 3 min, held at 67% B for 5 min, and then raised to 90% B until 12 min, held at 90% B until 15 min, decreased to 50% B over the next min, and held at 50% B until 20 min for re-equilibration of the system prior to the next injection. Solvent C was initially 5% and then lowered to 3% until 3 min, held at 3% until 8 min, raised to 5% until 12 min and then kept constant at 5% throughout the run. A flow rate of 0.25 mL/min was used, the column temperature was 30°C and the injection volume was 1 µL. MS acquisition was carried out with a heated electro spray ionization ion source operated in switching mode. The source parameters were similar for both negative and positive modes: sheath gas flow rate, auxiliary gas flow rate and sweep gas flow rate: 50, 20 and 0 arbitrary units respectively; capillary temperature: 350°C; heater temperature: 50°C; spray voltage: 3.00 kV. Ten-point of analytical standards were prepared in EtOH. The concentration range in CBD calibration curves was 0.1–125 ng/mL. In addition, calibration curves of 7-OH-CBD in the 0.125-100 ng/mL range and 7-COOH-CBD in the 1.25–100 ng/mL range were also prepared. All the calibration curves were determined according to the weighted least-squares linear regression method with a weighting factor of 1/X, R2 = 0.99. Area-under-the-curve (AUC) values were calculated directly from the serum concentration–time curve using the linear trapezoidal method for all eight timepoints.
2.6. Statistical Analysis
Non-compartmental analysis of PK parameters was performed using the PKSolver program (China Pharmaceutical University, Jiangsu, China) [51]. Data was determined as mean ± SEM, significant differences among experimental groups were determined using an unpaired student t-test with p < 0.05.
{kind=link}