Chemicals
Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS), 3isobutyl1methylxanthine (IBMX), penicillin and streptomycin, isopropanol, insulin, dexamethasone (DEXA) were procured from Thermo Scientifics, USA. MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) and Oil-Red-O (ORO) stain were procured from Sigma Aldrich, USA. Analytical Grades’ of other solvents, chemicals and reagents were to be utilized for the experiments.
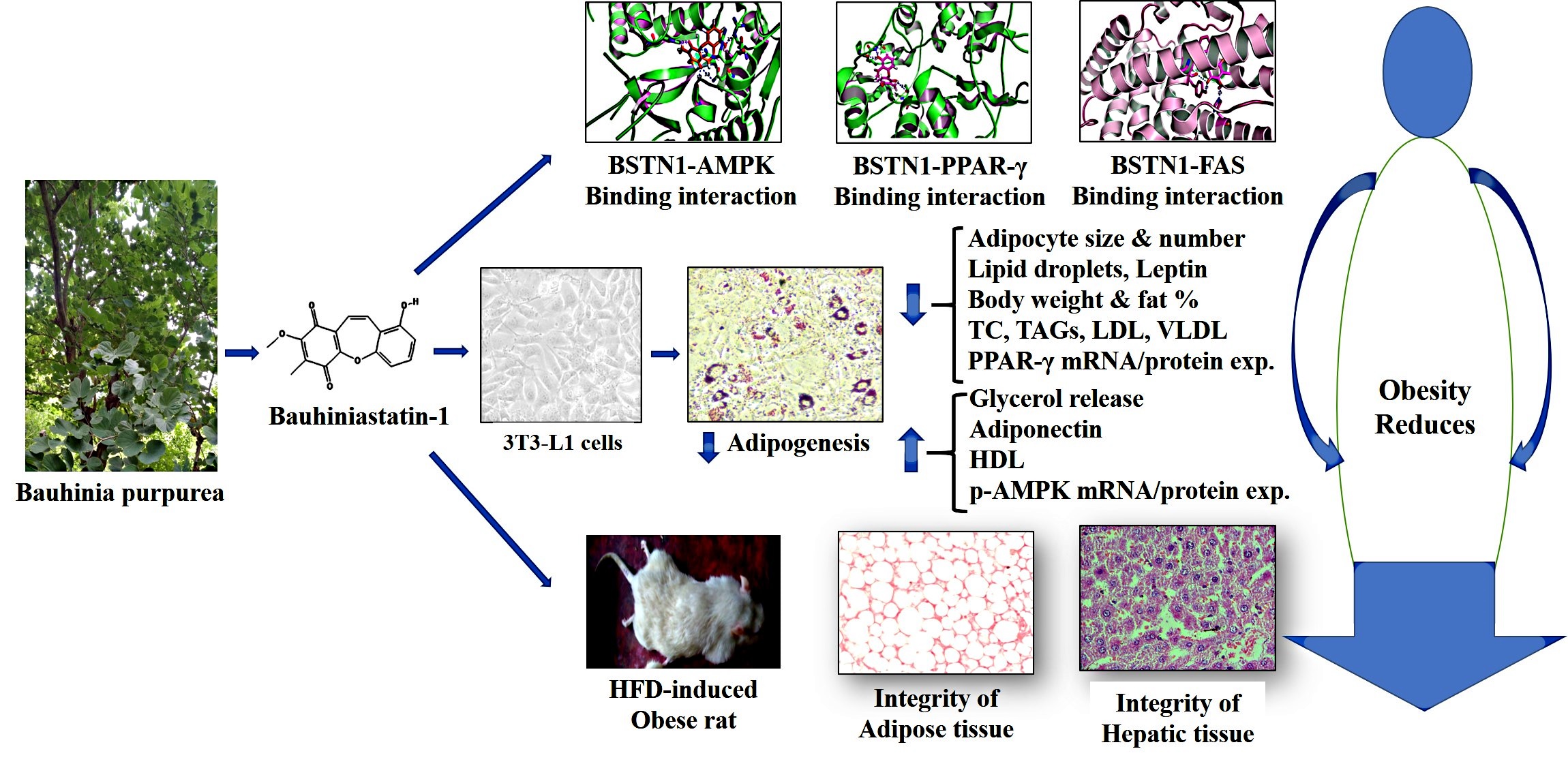
Isolation and purification of Bauhiniastatin-1 (BSTN1)
The bark of B. purpurea was collected from the Seshachalam forests, Tirupati, Andhra Pradesh, India. It is authenticated by taxonomist in the department of Botany, Sri Venkateswara University, Tirupati, voucher number is 136 and specimen was preserved in departmental herbarium. The bark of B. purpurea was powdered, extracted with ethanol and further fractionated by column chromatography using different solvents [12]. The collected fractions were subjected to LC-MS analysis on 6520 Accurate Q-TOF (Agilent Santa Clara, CA) mass spectrometer to identify the major compounds. Bauhiniastatin-1 was purified using an HPLC system equipped with a binary gradient system, a variable UV-VIS-detector and a Rheodyne Model 7725 injector with a loop size of 20 µL, and an integrator. Reverse phase chromatographic analysis was carried out in isocratic conditions using a C-18 reverse phase column (250 x 4.6 mm id., 5 µL C-18) at 40°C. Mobile phase consisted of methanol:water (20:80 v/v) with a flow rate of 1mL/min. The detection of compounds was performed at 220 nm. A single sharp peak at 5.942 min of retention time was identified as BSTN1 [13].
Cell culture and differentiation of adipocytes
The 3T3L1 pre-adipocytes of American Type Culture Collection (ATCC) cells were maintained and cultured in DMEM supplemented with 10% FBS at 37°C in a humidified atmosphere with 5% CO2. For adipogenesis studies, 3T3-L1 were grown to confluence, cells were stimulated with adipogenesis differentiation medium of induction (DMI) consisting of DMEM, 5% FBS, 0.5 mM IBMX, 1 µM DEXA and 10 µg/mL insulin for 2 days followed by treating cells with differentiation medium (DMEM with 5% FBS and 10 µg/mL insulin) for additional 8–10 days [14]. All the media that we used contained 100 IU/mL penicillin and 100 mg/mL streptomycin. A volume of 0.01 % DMSO was used as vehicle control for in-vitro experiments. For evaluating anti-adipogenic effects of BSTN1, 3T3-L1 cells cultured and adipogenesis was induced in 12 well plates with different concentrations of BSTN1 (5, 10 and 20 µM) in differentiation medium. The adipocytes were stained for neutral lipids (lipid droplets) and observed under a bright field microscope or used for other studies.
Cytotoxicity studies / Cell viability assay (MTT assay)
The 3T3L1 preadipocytes were cultured in DMEM and cell viability assays were conducted as previously described [15, 16].
Determination of adipocyte lipid content using quantitative Oil Red O staining
Lipid contents in adipocytes were visualized as well as measured using Oil Red O (ORO) staining [17, 18].
Lipolysis studies
The lipolysis studies were conducted by measuring glycerol levels released into the cell culture medium, using commercial kit (Lipolysis assay kit, ab185433, Abcam, Shangai) following manufacturer’s instructions [19]. The glycerol content was expressed as nmol/well.
RT-PCR studies
Total RNA was isolated from 3T3-L1 cells by using tri-reagent (Sigma Aldrich, USA) according to manufacturer’s protocol and reverse transcribed to obtain cDNA using cDNA synthesis kit (Applied Bio Systems, Foster City, USA) [20]. Two nano grams of cDNA was used for RT-PCR. The PCR amplification was performed with transcript specific primers (Additional file S1).
Animal studies
Male WNIN rats (aged 5–6 weeks), normal pellet diet and high fat diets were obtained from National Institute of Nutrition (NIN), Hyderabad, India. After one-week acclimatization period, rats were fed with either normal diet or HFD, water ad-libitum and maintained at standard laboratory conditions (temperature: 23°C ± 2°C; humidity: 40–60%) for 18 weeks as described in experimental design. Among different types of HFDs used to induce obesity, a HFD with 60% calories from fat induces obesity most effectively comparable to western diet [21, 22]. Animals were fed on 20% protein diet (normal pellet diet) (low fat control diet, percent of energy from carbohydrate-64%, protein-20% and fat-16%) (Additional file S2) or high fat diet (percent of energy from carbohydrate-20%, protein-20% and fat-60%) (Additional file S3) contained all the recommended macro and micronutrients. To test the therapeutic activity of BSTN1, 1.25, 2.5 and 5 mg/kg b.wt. of BSTN1 was suspended in 0.5% carboxymethylcellulose (CMC) and orally administered for 6 weeks from 13th week onwards using an intra-gastric tube. We selected these concentrations based on our initial pilot studies using solvent extracts [12].
IAEC approval (No:55/2012/(i)/a/CPCSEA/IAEC/SVU/MBJ, dated: 8-7-2012) were followed to conduct the animal experiments.
Experimental design
Rats initially weighing 160–180 g were randomly divided into six groups of six each (n = 6).
Group I: Normal pellet diet fed rats
Group II: HFD-fed rats (Placebo)
Group III: HFD fed + BSTN1 (1.25 mg/kg. b.wt./day) treated rats
Group IV: HFD fed + BSTN1 (2.5 mg/kg. b.wt./day) treated rats
Group V: HFD fed + BSTN1 (5 mg/kg. b.wt./day) treated rats
Group VI: HFD fed + Orlistat (5 mg/kg. b.wt./day) treated rats
Measurement of body weight and body composition parameters
The body composition, body weight, fat percent of each rat was measured by Total Body Electrical Conductivity (TOBEC) using a small animal body composition analysis system (EM-SCAN, Model SA-3000 Multi detector, Springfield, USA). At the end of the experiment, animals were anesthetized using isoflurane, blood was collected by heart puncture method. Plasma and/or serum were separated by centrifugation at 2500 rpm for 15 min. Various organs and tissues including abdominal adipose tissue and liver were dissected, and stored appropriately. For histology studies tissues were fixed and processed as described in later sections.
Plasma leptin and adiponectin levels
Plasma leptin and adiponectin are key adipokines secreted by adipocytes. Adipokine levels were measured in experimental rats using enzyme-linked immunosorbent assay kits (Crystal Chem, Downers Grove, IL, USA). These assays were performed in duplicates (n = 6), as per the manufacturer’s guidelines and adipokine levels were expressed in ng/mL [23].
Estimation of serum lipid profile
Serum total cholesterol (TC) was estimated by CHOD-PAP method, triglycerides (TGs) was estimated by GPO-TOPS method, HDL-cholesterol, VLDL-cholesterol, LDL-cholesterol were estimated by selective inhibition method (Agappe Diagnostics Ltd., Kerala, India), phospholipids (PLs) and free fatty acids (FFAs) were assessed as previously described [24].
Estimation of hepatic lipid levels
Lipids were extracted from the livers of experimental animals as described [25]. In brief, the tissues were rinsed with ice-cold physiological saline, homogenized in cold chloroform-methanol (2:1, v/v) and the contents were extracted for 24 h. The extraction was repeated four times. The combined filtrate was washed with 0.7% potassium chloride and the aqueous layer was discarded. The organic layer was made up to a known volume with chloroform and used for hepatic lipid analysis.
Measurement of AST and ALT activities
Hepatic marker enzymes, aspartate transaminase (AST) and alanine transaminase (ALT) activities were estimated at the end of the experiment by using commercially available kit (Agappe Diagnostics Ltd, Kerala, India) following the manufacturer's protocol.
Western blot analysis
Adipose and hepatic tissue proteins were extracted with lysis buffer (Sigma Aldrich, USA) and quantified using Bradford method [26]. Equal amount of protein (40 µg) was resolved on 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane. To block non-specific binding sites, blots were incubated at room temperature with 5% skimmed milk (v/v) for 1 h followed by overnight incubation with primary antibodies of anti-PPAR-γ, anti-AMPK and mouse anti-β actin (1:1,000 dilution, ABclonal Technology, USA) at 4°C. The immuno-reactive antigen was then recognized by incubation with HRP-conjugated secondary antibody (1:1,000 dilution, Abclonal Technology, USA). Immuno-reactive bands were visualized using chemiluminescence detection system.
Histopathological examination
Adipose and liver tissues were collected from both control and experimental rats and fixed in formalin solution. A small piece of tissue was cut, trimmed, processed and prepared paraffin blocks. Then the paraffin blocks were sectioned (5–8 µm) using microtome and stained using haematoxylin and eosin (H&E) following standard histology protocol [27].
Molecular docking studies (Accession of target protein)
The three-dimensional structure of FAS (6NNA), AMPK (6C9F), PPAR-γ (3WMH) and BSTN1 were downloaded from the RCSB protein Data Bank and Pub chem. The atomic coordinates of the ligand were geometrically optimized using Argus Lab 4.0.1. [28]. In-silico studies were carried against FAS (6NNA), AMPK (6C9F) and PPAR-γ (3WMH), with ligand (BSTN1) using the docking program Patchdock [29]. After the docking, protein–ligand complexes were studied using PyMol viewer tool (www.pymol.org)1. Protein and ligand interactions were analysed and visualized through PyMol viewer tool (www.pymol.org)1.
Statistical analysis
The results are expressed as the mean ± standard deviation (SD), and comparison was made by using one-way ANOVA programme followed by Tukey’s post hoc tests to study the significance level (SPSS, version 17.0; SPSS Inc., Chicago, IL, USA).
{kind=link}