2.1. Synthesis and characterization:
Starting materials, and solvents were purchased from commercial sources and used without further purification.
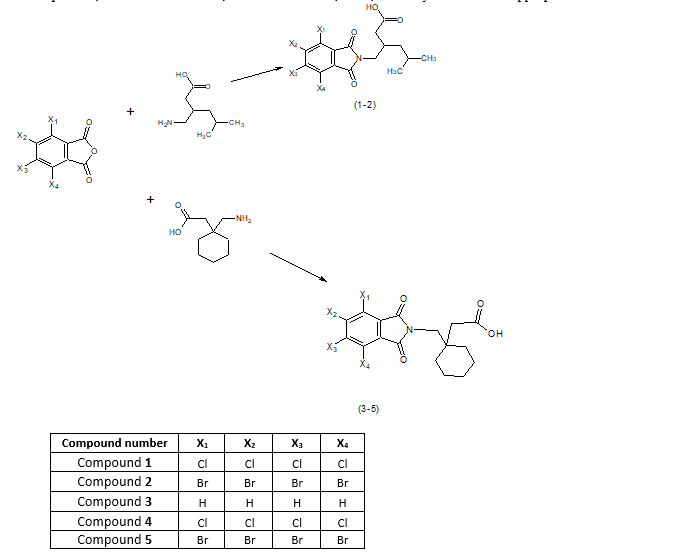
Scheme 1 shows synthesis reactions, we used two procedures, procedure A(26), and procedure B(27). Progress of reactions were monitored by TLC on silica-gel plates (Merck 60F254).
Procedure A
0.010 mole of phthalic anhydride or its derivatives, and 0.011 mole of pregabalin (1–2) or Gabapentin (3–5) were dissolved in 5 mL dimethyl form amide (DMF) in a round bottom flask. Solution was refluxed in oil path at temperature 180 Co, completion of reaction was determined by TLC. Reaction mixture was poured into ice cold water, crude product was precipitated, filtered out on a Buchner funnel, washed with water, dried, and purified by recrystallization from appropriate solvents.
Procedure B
Equimolar quantities of phthalic anhydride or its derivatives, and pregabalin (1–2) or Gabapentin (3–5), were grounded together in a mortar for 1 minute, then mixture was transferred to glass beaker containing a magnetic stirrer. Few drops of DMF were added to the beaker, and it was irradiated with microwaves (750 W) with continuous stirring for 5 minutes. completion of reaction was determined by TLC.
Mixture was cooled to room temperature, 50 mL ice-cold water was poured into the flask to precipitate the crude product, and it was filtered off, washed with water, dried, and recrystallized from appropriate solvent.
Melting points were determined using Stuart melting point apparatus SMP30 and used without calibration. High performance liquid chromatography was performed using Shimadzu UFLC apparatus A20.
IR spectra were recorded in KBr on Thermo Nicolet 6700 FT-IR spectrophotometer apparatus. UV spectra were recorded using JENWAY 6850 UV/Vis spectrophotometer. 1H-NMR and 13C-NMR spectra were recorded on a Bruker Ultra Shield 400 Instrument (400 MHz for 1H, 100 MHz for 13C).
ESI–MS experiments were performed using Agilent 6420 Triple Quadrupole LC/MS apparatus equipped with a standard ESI source. Instrument was operated in the positive-ion mode. Spectra were recorded for samples dissolved in DMSO: H2O.
5-methyl-3-[(4,5,6,7-tetrachloro-1,3-dioxo-1,3-dihydro-2H-isoindole-2-yl) methyl] hexanoic acid (1):
Pale yellow crystals, yield 90% (procedure A)-92% (procedure B), MP 177.6–180 CO, UV (methanol, nm): λmax 288, 333.
IR (KBr, cm− 1):3330.2 (OH), 2954.9, 2870 (CH2 stretching), 1772.4 (CO), 1732.4 (CO), 1702.5 (CO), 1437(CH2 bending), 1165 (C-N).
1H NMR (400 MHz, DMSO-d6, ppm): 12.009(s,1H, OH), 3.6–3.42 (m, 2H, N-CH2), 2.18–2.35 (m,2H, CH2-COOH), 2.18–2.048 (m,1H,N-CH2-CH), 1.793–1.634 (m,1H,CH (CH3)2), 1.0785–1.267 (m,2H,CH2-CH(CH3)2), 0.7587–1.0058 (2d, 6H, (CH3)2).
13C NMR (100 MHz, DMSO-d6, ppm): 174.037 (COOH), 164.175 (2CO), 138.474 (2aromatic carbons), 128.887(2 aromatic carbons), 128,454 (2 aromatic carbons), 42.637, 41.572, 37.682, 32.157, 25.198, 23.207, 22.677.
DEPT 135 13C NMR (100 MHz, DMSO-d6, ppm): 42.592 (N-CH2), 41.547 (CH2-COOH), 37.659 (CH2CH (CH3)2), 32.135(N-CH2-CH-), 25.185 (CH (CH3)2), 23.223 (CH3), 22.664 (CH3).
ESI+ MS-MS (M/Z): 101.0, 240.8, 285.9, 287.9, 289.9, 291.9, 307.8, 309.9, 311.9, 362.1.
5-methyl-3-[(4,5,6,7-tetrabromo-1,3-dioxo-1,3-dihydro-2H-isoindole-2-yl) methyl] hexanoic acid (2):
White crystals and crystalline powder, yield: 83% (procedure A)-86% (procedure B), MP 212-212.6 CO .
UV λmax (methanol, nm): 288, 338. IR(KBr,cm− 1):3399.5 (OH), 2954.6, 2869.6 (CH2 stretching), 1769.9 (CO), 1732.3 (CO), 1703.4 (CO), 1610.7 (C = C arom.), 1561.7 (C = C arom.), 1435 (CH2 bending), 1163.5 (C-N).
1H NMR (DMSO-d6, 400 MHz, ppm): 11.995(s,1H, OH), 3.6–3.42 (m, 2H, N-CH2), 2.18–2.35 (m,2H, CH2-COOH), 2.18-2.0414 (m,1H, N-CH2-CH), 1.776–1.636 (m,1H, CH(CH3)2), 1.059–1.216 (m,2H, CH2-CH(CH3)2), 0.790–0.994 (2d, 6H, (CH3)2).
13C NMR (DMSO-d6, 100MHz, ppm): 174.074 (COOH), 164.493 (2CO), 136.463 (2aromatic carbons), 131.614(2 aromatic carbons), 120.831 (2 aromatic carbons), 42.796 (N-CH2), 41.632(CH2COOH), 37.762((CH2-CH(CH3)2), 32.083(N-CH2-CH), 25.215(CH(CH3)2), 23.250(CH3), 22.652(CH3).
DEPT 135 13C NMR: 42.796 (N-CH2), 41.632(CH2COOH), 37.762((CH2-CH(CH3)2), 32.083(N-CH2-CH), 25.215(CH(CH3)2), 23.250(CH3), 22.652(CH3).
ESI+MS-MS (M/Z): 679.8 [M + DMSO + H]+, 604.7 [M]+, 588.7 [M-OH]+, 565.7,476.7, 463.7.
{1-[(1,3-dioxo-1,3-dihydro-2H-isoindole-2-yl) methyl] cyclohexyl} acetic acid (3):
White fine powder, yield:80% (procedure A)-84% (procedure B), MP 151–152 CO, UV λmax (methanol, nm):290, 302.
IR(KBr, cm− 1): 3434 (OH), 3096 (Ar-H stretch.), 2929, 2869 (CH2 stretching), 1770 (CO), 1720 (CO), 1710 (CO), 1611 (C = C arom), 1466 (CH2 bending), 1195 (C-N).
1H NMR (400 MHz, CD3CN, ppm): 11.99(s,1H, OH), 7.91–7.78 (m, Ar-4H), 3.654(s,2H, N-CH2), 2.304(s,2H, CH2-COOH), 1.670–1.108 (m, 10H of cyclohexane).
13C NMR (100 MHz, CD3CN, ppm): 173.287(COOH), 169.243 (2CO), 134.784(2 aromatic carbons), 132.131(2 aromatic carbons), 123.454 (2 aromatic carbons), 46.146, 38.098, 33.365, 25.768, 21.551.
DEPT 135 13C NMR: 134.787 (2 aromatic carbons), 123.459 (2 aromatic carbons), 46.141 (CH2-N), 38.098 (CH2-COOH), 33.349 (2(CH2) of cyclohexane), 25.767(CH2 of cyclohexane), 21.546(2 (CH2) of cyclohexane).
ESI+MS-MS (M/Z): 302.0 [M + H] +, 323.9 [M + Na] +, 339.9 [M + K] +, 284.0[M-OH] +, 256.1[M-COOH] +, 242 [M-(CH2-COOH)] +, 160[M-(COOH-CH2-C6H10)] +.
{1-[(4,5,6,7-tetrachloro-1,3-dioxo-1,3-dihydro-2H-isoindole-2-yl) methyl] cyclohexyl} acetic acid (4):
White crystals yield:70% (procedure A)-75% (procedure B), MP 218-218.4 CO, UV λ max (methanol, nm):202, 212, 236, 333.
IR(KBr, cm1): 3299 (OH), 2862, 2931 (CH2 stretching), 1772 (CO), 1733 (CO), 1703 (CO), 1434 (CH2 bending), 1157 (C-N), 736 (C-Cl).
1H NMR (400 MHz, DMSO-d6, ppm): 12.031(S,1H, OH), 3.662 (S,2H, N-CH2), 2.323 (s,2H, CH2-COOH), 1.618–1.139 (m, 10H of cyclohexane).
13C NMR (DMSO-d6, 100 MHz, ppm): 173.281(COOH), 164.820 (2CO), 138.412(2 aromatic carbons), 128.975(2 aromatic carbons), 128.375 (2 aromatic carbons), 46.954, 38.212, 33.144, 25.668, 21.533.
DEPT135 (DMSO-d6, 100 MHz, ppm): 46.948 (CH2-N), 38.212 (CH2COOH), 33.144 (2(CH2) of cyclohexane), 25.690 (CH2 of cyclohexane), 21.526(2(CH2) of cyclohexane).
ESI+MS-MS (M/Z):407.9, 497.8, 387.8, 301.
{1-[(4,5,6,7-tetrabromo-1,3-dioxo-1,3-dihydro-2H-isoindole-2-yl)methyl]cyclohexyl}acetic acid (5):
Pale yellow crystals yield: 77% (procedure A)-80% (procedure B), MP 241.5–243 CO, UV λ max (methanol, nm):287, 336.
IR(KBr, cm1): 3299 (OH), 2929, 2861 (CH2 stretching), 1770 (CO), 1735 (CO), 1702 (CO), 1559 (C = C),1432 (CH2 bending), 1155 (C-N), 669 (C-Br).
1H NMR (400 MHz, DMSO-d6, ppm): 12.004(S,1H, OH), 3.645 (S, 2H, N-CH2), 2.308 (S,2H, CH2-COOH), 1.72–1.07 (m, 10H of cyclohexane).
13C NMR (DMSO-d6, 100 MHz, ppm): 173.308(COOH), 165.135 (2CO), 136.588(2 aromatic carbons), 131.689(2 aromatic carbons), 120.784 (2 aromatic carbons), 47.112(N-CH2), 38.192 (CH2-COOH), 33.171(2C of cyclohexane), 25.665 (1C of cyclohexane), 21.555 (2C of cyclohexane).
DEPT135-13C NMR (DMSO-d6, 100 MHz, ppm): 47.059 (CH2-N), 38.361 (CH2-COOH), 33.226 (2(CH2)), 25.778 (CH2), 21.547 (2(CH2)).
ESI+MS-MS (M/Z):588.3, 566.3, 475.2,452.6.
2.2. Evaluation of biological activity:
2.2.1. Antioxidant activity:
Antioxidant activity of prepared compounds was determined by measuring free radical scavenging activity using DPPH (2,2-diphenyl-1-picrylhydrazyl) according to standard method, and Ascorbic acid was used as positive control(28).
We have studied serial concentrations 250, 500, 750, and 1000 µg/mL of each compound, and Ascorbic acid in methanol.
1 mL of studied solution was mixed with 3 mL of DPPH solution, and allowed to stand in dark for 30, 60, 90, 120 minutes.
Methanol was used to prepare negative control. Pure methanol was used as a blank. Absorbance was measured at 517 nm after 30, 60, 90 and 120 minutes. All experiments were done in triplicates and the values are averaged.
DPPH scavenging activity was calculated though following equation:
Scavenging effect % = (Anegative control - Asample/Anegative control) x 100
IC50 was calculated from concentration-inhibition curve by plotting the sample concentration versus the corresponding DPPH scavenging activity.
2.2.2. Antimicrobial activity:
Antimicrobial activity of parent compounds (Pregabalin, and Gabapentin) and synthesized compounds was investigated against gram positive (Staphylococcus aureus) and gram negative (Escherichia Coli) bacteria by well diffusion method reported by Perez et al(29).
Stock solution of each compound was prepared, and diluted to prepare other concentrations through two-fold dilution method. At least 6 concentrations were studied for each compound (3200, 1600, 800, 400, 200, 100 µg/mL).
Microbial suspension was prepared in physiological saline serum, and adjusted to 0.5 McFarland standard.
Culture media was prepared by pour plate method, in which microbial suspension was added to Muller Hinton Agar at percentage 1%, then poured in petri dishes (depth 3–4 mm).
Poured Plates were left to cool then similar wells (8 mm in diameter) were made in the agar and loaded with 100 µl of the tested compound solution. Inculcated plates were incubated at 37°C for 24 h.
Antimicrobial activity was evaluated by measuring the zone of inhibition (IZ) in mm. Each experiment was carried out in triplicate and the average zone of inhibition was calculated.
Gentamycin sulfate was used as standard drug for antibacterial activity, pure solvent was used as negative control.
2.2.3. Anticancer activity:
Effect of compounds was studied on human cancer cell lines Caco-2 and HCT-116 (Sigma Aldrich).
Stock solution of each compound was prepared in DMSO, and diluted with culture medium to prepare serial concentrations. The final concentration of DMSO in wells is less or equal to 0.1%.
Viability was measured after 48 hours using XTT method, cell cycle changes of treated cells were studied by Flow Cytometry, Assay of apoptosis and necrosis cell death was performed by Annexin V/propidium iodide (PI) double staining assay method using Flow Cytometry.
2.2.3.1. Cell cultures:
Human cancer cell lines Caco-2 and HCT-116 were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 100 U/mL penicillin and 100 µg/mL streptomycin in sterile flasks. The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
2.2.3.2. XTT assay:
Human Caco-2 and HCT-116 cultures were seeded in 96-well plates (1.5x104 cells/well).
Plates were incubated for 24 hours at 37°C and humidified atmosphere containing 5% CO2, then culture medium was removed and cells were treated with different concentrations of studied compounds.
At least six concentrations were studied for each compound, and each concentration was studied in three triplicates. DMSO alone was added to a set of wells as the solvent control, culture medium was added to another set of wells as negative control.
After treatment, plates were incubated for 48 hours at temperature 37 CO and 5% CO2 atmosphere.
Antiproliferative effect was measured after 48 hours by XTT assay reported by Skehan et al.(30) according to instructions of manufacturing company of the Kit (Roche).
Absorbance was measured using ELISA reader spectrophotometer at 450 nm. All experiments were carried out in triplicates.
Inhibition of cell viability was expressed as percentage and it was calculated as following:
Growth inhibition GI%= [1- (absorbance of test compound/absorbance of the negative control)]*100
Inhibition ratio was calculated as mean of results of all replicates. IC50 was calculated from dose- response curve.
2.2.3.3. Cell Cycle analysis:
We have studied cell cycle phases in treated cells, compared with untreated cells, in order to get more information about the mechanism of anticancer effect of synthesized compounds.
Cells were cultured and treated with studied compounds for 72 hours, untreated cells were incubated with culture medium in the same conditions as negative control.
Cells were harvested by trypsin, samples were centrifuged, cellular precipitate was washed with phosphate-buffered saline (PBS), and resuspended in RPMI1640.
Samples were fixed by incubation with methanol in dark place for 30 minutes at -20 Co.
Samples were centrifuged, and fixed cells were rinsed twice with PBS. Staining solution (DNA fluorochrome PI in a solution containing Triton X-100 and RNase) was added, and samples were kept in dark for 30 minutes at 4–8 CO. Stained cells were analyzed by BD FACS Calibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) in accordance with the manufacturer's protocol (31).
The tests were performed in duplicates and repeated at least twice.
2.2.3.4. Apoptosis assay:
Assay of apoptosis and necrosis cell death was executed by Annexin V/propidium iodide (PI) double staining assay method in order to know effect of compounds on cell death.
Cells were treated with compounds for 72 h, untreated cells were incubated with RPMI1640 as a negative control.
Cells were harvested by trypsin, and precipitated by centrifuge. cellular precipitate was washed with PBS, and resuspended in culture medium.
Cells were fixed by incubation with methanol in dark and cold place for 30 minutes at -20 Co.
Fixed cells were separated by centrifuge, and rinsed twice by PBS.
Cells were stained with 5 µL Annexin V and 5 µL PI and 350 µL Annexin binding buffer, and analyzed by BD FACS Calibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Number of cells analyzed for each sample was 104 cells(31). Gating was implemented on the basis of negativecontrol staining profiles.
The tests were performed in duplicates and repeated at least twice.
{kind=link}