Structure and composition
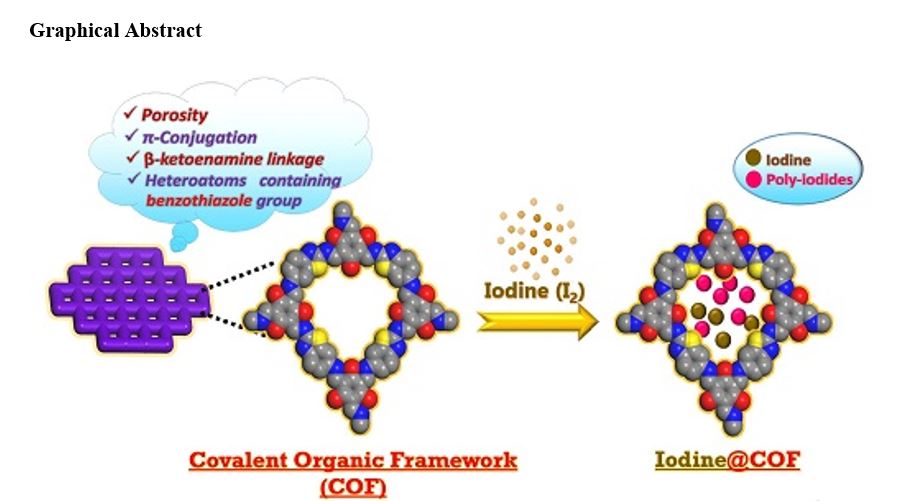
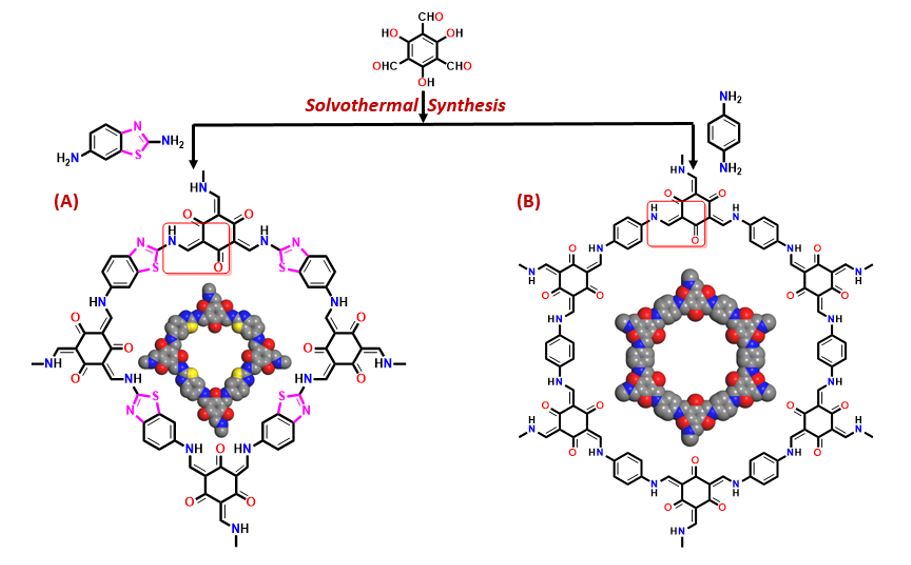
β-Ketoenamine-linked COF-3 and COF-4 were synthesized via the solvothermal condensation of 2,6-diaminobenzothiazole,1,4-phenylenediamine, respectively with 2,4,6-triformylphloroglucinol as depicted in Scheme 1. The synthesized COFs were characterized using ATR-Infra-red (IR), solid-state NMR spectroscopy, powder X-ray diffraction (PXRD), FE-Scanning Electron Microscopy (FE-SEM), high resolution Transmission Electron Microscopy (HR-TEM), X-ray Photoelectron Spectroscopy (XPS) and Thermogravimetric analysis (TGA).
IR spectra of the two COFs revealed the disappearance of characteristic bands of the starting materials i.e. N-H stretching (3100–3400 cm− 1) and aldehyde carbonyl stretching (1645 cm− 1) bands, that confirmed the complete Schiff base condensation reaction between the monomers (Figs. 1 & 2) [60]. Both the COFs showed β-ketoenamine linkage in their structures as evident from the absence of O-H stretching and C = N stretching bands, the characteristic bands for the structure to exist in the enol form. The tautomerism between the β-positioned hydroxyl groups and Schiff bonds (C = N) leads to the intramolecular NH-O hydrogen bonding resulting into the β-ketoenamine linkages. Also, the appearance of C-N stretching peaks at 1262 cm− 1 and 1259 cm− 1, the characteristic peak of β-ketoenamine linkage in case of COF-3 and COF-4, respectively was also observed. No C = O stretching band corresponding to keto group of the framework was observed in the IR-spectra of both the COFs. This is because C = O stretching band merged with the C = C stretching band at 1588 cm− 1 and 1582 cm− 1 in the extended structures both the COFs viz. COF-3 and COF-4, respectively.
The atomic-level construction of both the COFs was further verified by 13C cross-polarization magic angle-spinning (CP-MAS) solid-state carbon NMR spectroscopy. NMR spectrum of COF-3 showed the signals at 185.80 ppm corresponding to carbonyl group of the β-ketoenamine linkage in the framework (Fig. S4). The signals at 148.53 ppm, 133.19 ppm, 121.42 ppm and 106.28 ppm correspond to aromatic carbons. The signals at 171.19 ppm and 161.35 ppm were attributed to the presence of thiazole in the structure of COF-3. Also, COF-4 showed signals at 106.32 ppm, 115.04 ppm, 121.85 ppm, 135.69 ppm and 183.78 ppm corresponding to aromatic carbons and carbonyl group of β-ketoenamine linkage, respectively (Fig. S5).
The powder X-ray diffraction patterns of the synthesized COFs showed their semi-crystalline nature. COF-3 displayed diffraction peak at 26.28° corresponding to the (002) reflection plane and COF-4 exhibited the major diffraction peak at 26.12° corresponding to the reflection from (201) plane. The π − π stacking distances between the COF-3 layers was 3.4 Å from the d spacing between (002) planes and in case of COF-4, the d-spacing between (201) reflection planes was 3.3 Å. To analyse the structures of the COFs and to calculate the parameters of the unit cell, geometrical optimizations and structural models were employed using Material Studio software 8.0. The experimental patterns of COF-3 matched well with the simulated staggered AB stacking model corresponding to the tetragonal lattice with space group P4222. Also, the experimental diffraction patterns of COF-4 matched well with simulated eclipsed AA stacking model corresponding to hexagonal lattice with space group P6/m. The lattice parameters of the unit cell were refined using Pawley refinement tool which gave the values for COF-3 as a = b = 3.13 Å, c = 6.81 Å, α = β = γ = 90° with Rwp = 6.09% and Rp = 4.84% for COF-3. Similarly, COF-4, exhibited the parameters as a = b = 22.91 Å, c = 3.34 Å, α = β = 90°, γ = 120° with Rwp = 5.27% and Rp = 4.16% for COF-4. (Figs. 3 & 4).
The surface areas of the COFs were further determined by nitrogen adsorption-desorption studies. Based on the nitrogen sorption isotherms, COF-3 and COF-4 were estimated to have Brunauer- Emmett-Teller (BET) surface areas of 57.9 and 133.9 m2/g, respectively as shown in Fig. 5. From isotherm curves, both the COFs were found to show type-III adsorption. Pore calculations were performed using Barrett-Joyner-Halenda (BJH) plots. The pore sizes for COF-3 and COF-4 were calculated to be 1.9 nm and 1.5 nm respectively and the pore volumes were 0.305 cc/g and 0.937 cc/g.
FE-Scanning electron microscopy (FE-SEM) images revealed coral like morphology for both the COFs in the microscopic range and pores could be seen on magnifying the images to the nano scale range. However, COF-4 seems visibly more porous than COF-3 (Fig. 6).
HR-TEM analysis of COF-3 and COF-4 also revealed the porosity in the frameworks with the stacking between the COF-sheets (Fig. S6).
Thermogravimetric analysis (TGA) of COFs determined the thermal stabilities of both the frameworks from 30°C to 600°C. COF-3 was thermally stable up to 210°C with ~ 10% weight loss in the beginning up to 75°C plausibly because of the guest solvent molecules within the pores of the frameworks. After 210°C, there was gradual weight loss up to 440°C followed by sharp loss up to (Fig. S7). COF-4 also showed 18% loss of the weight at 70°C plausibly due to the guest solvent molecules and then, thermal stability up to 280°C, a gradual weight loss up to 400°C followed by a sharp weight loss (Fig. S7).
XPS analysis of COF-3 revealed the binding energies at 532.2 eV, 398.6 eV and 164.1 eV corresponding to the O1S, N1S and S2p orbitals related to carbonyl, C-N and C-S bonds. In case of COF-4, the binding energies corresponding to carbonyl group and C-N bond were present suggesting the formation of enamine linkage within the frameworks. (Fig. S8)
Iodine Adsorption Studies
Adsorption of iodine vapor
Adsorption of iodine vapors was studied by exposing an appropriate amount of COF (40 mg) in a closed vessel to the iodine vapors at 77°C and ambient pressure to have the close scenario of typical nuclear fuel reprocessing conditions. During adsorption process, the deepening of the COF color from light brown to dark brown affirmed the diffusion of iodine into pores of the frameworks. COF-3 after adsorbing iodine was referred as I2@COF-3. The iodine uptake capacity reached 0.725 g g− 1 after 23 hours and reached the equilibrium value 1.075 g g− 1 after three days (Fig. 7; Table 1).
Table 1
Parameters and iodine capacity of BT-COF.
| COF | Pore topology | Pore size (nm) | BET surface area (m2g− 1) | Pore volume (cm3g− 1) | Theoretical capacity (g g− 1) | Experimental capacity (g g− 1) | Pore accessibility (%) |
| BT-COF | Tetragonal | 1.9 | 57.9 | 0.305 | 1.51 | 1.08 | 71.5 |
Iodine adsorption from solution
The adsorption of iodine from n-hexane solution was performed at various concentrations viz. 50, 100, 150, 200, 250 mg L− 1 under ambient temperature and pressure conditions. The UV-Vis spectrum of iodine solution after adsorption at various intervals of time is shown in Fig. 8 for 250 mg L− 1 concentration. Adsorption of the iodine solution with 250 mg L− 1 concentration increased significantly in the first one hour and then it reached the equilibrium value after 270 minutes. The removal efficiencies reached 43.01% and 65.9% after 4.5 hours for COF-3 and COF-4, respectively with the initial concentration of 250 mg L− 1. The adsorption kinetics was studied using pseudo-first order and pseudo-second order kinetic models. The experimental data for COF-3 fitted well with both pseudo-first order (R2 = 0.9868) and pseudo-second order (R2 = 0.992) kinetics with R2 values close to each other. This suggests the involvement of both physical and chemical adsorptions. The pseudo-first and pseudo second order rate constants were calculated to be 0.0134 min− 1 and 8.9 x 10− 6 min− 1 for COF-3. However, the equilibrium adsorption capacities calculated from both the kinetics curves are compared with the experimental capacity, pseudo-first order curve fitted comparable to pseudo-second order curve. In case of COF-4, pseudo-first order kinetics (R2 = 0.9337) fitted well with the experimental data than the pseudo-second order (R2 = 0.8747) kinetics. This pointed towards the physical interaction between iodine molecules and the frameworks of COF-4. The calculated pseudo first and second order rate constants were calculated to be 0.0054 min− 1 and 5.1 x 10− 5 min− 1(Fig. 9; Table 2).
Further, adsorption isotherms were evaluated using Langmuir and Freundlich isotherm models. Both Langmuir and Freundlich models fitted well the sorption curves, with a closely related correlation coefficient (R2) of 0.9976 and 0.9899, respectively for COF-3 suggesting the formation of multilayer during the adsorption of iodine. The calculated Langmuir adsorption constant (KF) was 0.0026 and maximum adsorption capacity from Langmuir model came out to be 135.1 mg g− 1. Also, Freundlich constant (KL) value came out to be 10.42 and n value to be 2.06. The adsorption isotherms of the COF-4 fitted well with the Freundlich model with the correlation coefficient (R2) of 0.9771. The Freundlich constant (KF) and n value came out to be1.68 and 1.04, respectively. Langmuir adsorption isotherm did not fit well with the sorption curve as the calculated maximum adsorption (555.5 mg g− 1) was much higher than the experimental value (143.0 mg g− 1) although correlation coefficient (R2) value (0.9817) was closer to that for COF-3. This suggested that multilayer adsorption dominated over monolayer adsorption (Fig. 10; Table 3).
Table 2
Kinetic parameters for the iodine vapor adsorption on COFs.
| COFs | BET surface area (m2 g− 1) | Conc. of solution (ppm) | Experimental qe (mg g− 1) | Pseudo first order | Pseudo second order |
| K1 (min− 1) | qe (mg g− 1) | R2 | K2 (min− 1) | qe (mg g− 1) | R2 |
| COF-3 | 57.9 | 250 | 109.0 | 0.0134 | 109.5 | 0.9868 | 8.9 x 10− 6 | 140.9 | 0.992 |
| COF-4 | 133.9 | 250 | 149.5 | 0.0054 | 132.1 | 0.9337 | 5.1 x 10− 5 | 188.7 | 0.875 |
Table 3
Adsorption isotherm parameters for Iodine adsorption from n-hexane solution
| COF | qm(exp) (mg g− 1) | Langmuir | Freundlich |
| KL | qm (mg g− 1) | RL2 | KF | n | RF2 |
| COF-3 | 109.0 | 0.0026 | 135.1 | 0.9976 | 10.42 | 2.06 | 0.9899 |
| COF-4 | 149.5 | 0.003 | 555.5 | 0.9817 | 1.68 | 1.04 | 0.9771 |
The mechanism of the iodine adsorption was preliminarily studied by Infrared spectroscopy. It was found that the characteristic peak positions of the frameworks changed significantly before and after the adsorption suggesting the charge transfer interactions between COF structures and the iodine molecules. There were shifts in the bands of C = O, C-N from 1586 cm− 1 to 1574 cm− 1and 1228 − 1220 cm− 1 in case of COF-3 and from 1579 cm− 1 to 1569 cm− 1,1251 to 1239 in case of COF-4. This shift in the bands suggested that adsorption of iodine molecules occurred at the enamine linkages (Fig. 11). Also, adsorption of iodine within the frameworks was supported by the FE-SEM a (Fig. 12). Both the pristine COFs showed nanowire like morphology with visible porosity. The observed porosities in case of the pristine COFs were reduced upon exposure to iodine molecules suggesting the adsorption of iodine molecules within the pores of the frameworks resulting into the reduced porosity of the frameworks.
{kind=link}
{kind=link}