Clinical Features
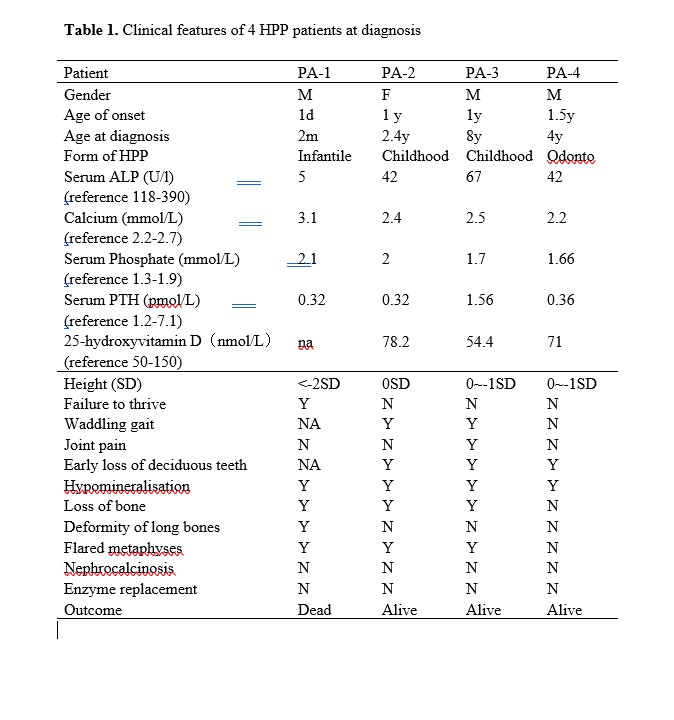
The clinical phenotypes of all four patients with HPP (three males and one female) from four unrelated families are summarized in Table 1 and Figure 1. They were all born to nonconsanguineous parents. They were initially referred to our clinic due to variable clinical manifestations, including growth failure, premature loss of teeth and rachitic symptoms. With biochemical tests, all patients showed remarkably decreased levels of ALP activity (Table 1). All patients were then suspected and finally diagnosed as HPP with confirmation of disease-causing mutations in the ALPL gene.
Patient 1 (PA-1) was delivered at term by c-section with a birth weight of 3000 g. He experienced respiratory distress and newborn pneumonia soon after he was born. He presented with feeding difficulty, seizure, poor head control and failure to thrive during the first two months of his life. He showed underweight, short stature, enlargement of the anterior fontanelle, and developmental delay when he was diagnosed at 2 months old. His serum ALP activity was almost undetectable (Table 1). X-ray showed general hypomineralization, deformity of long bones, flared metaphyses and hypolucent mid-metaphyses (Figure 1. A and B). He died of pneumonia and respiratory failure at 3 months old. Patient 1 was classified as infantile HPP (Table 1).
Patient 2 (PA-2) began to present premature loss of the deciduous teeth and muscle weakness at 1 year old. She had waddling gait and delay of walking. She had lost most of her deciduous teeth and only reserved seven teeth when she came to the clinic at 2 years old (Figure 1. C and D). Very low level of serum ALP activity (42U/L) suggested the diagnosis of HPP (Table 1). Panoramic radiographs showed premature loss of most of deciduous teeth, taurodontism in the deciduous molars and maldevelopment of permanent teeth (Figure 1. E). X-ray of wrist and knee revealed flared metaphyses and bone destruction in distal femur and proximal tibia (Figure 1. F and G). Patient 2 was diagnosed as childhood HPP at 2 years and 5 months old (Table 1).
Patient 3 (PA-3) began to present joint swelling, bone pain and muscle weakness at 1 year old. He experienced delay of walking and early loss of deciduous teeth. His symptoms of joint swelling and bone pain were not relieved at 8 years old. He had body weight and height within normal ranges. Serum ALP activity was 41 U/L at 4 years old and 67 U/L at 8 years old. Panoramic X-ray showed taurodontism, reduced alveolar bone, and enlarged pulp chambers and root canals (Figure 1. J). The X-ray revealed hypomineralization at 4 years old (Figure 1. H, I) and bone destruction in the proximal humerus at 8 years old (Figure 1. K, L). Patient 3 was diagnosed as childhood HPP at 8 years old (Table 1).
Patient 4 (PA-4) experienced premature loss of deciduous teeth since he was one and half years old. The physical examination was unremarkable, other than the absence of the upper and lower anterior incisor and canine teeth (Figure 1. M). Serum ALP activity was 42 U/L at diagnosis when he was four years old. Panoramic X-ray showed a reduced alveolar bone of whole dentition (Figure 1. N). His wrist and knee X-ray revealed normal growth plates without any evidence of defective bones (Figure 1. O and P). Patient 4 was diagnosed as odonto HPP at 4 years old (Table 1).
Mutational analysis
In order to confirm the diagnosis of HPP and correlate the phenotype with specific genotype, mutational analyses of the ALPL gene were performed in all four patients and their parents except the mother of patient 1 was not involved in this study. The ALPL variants in patient 1 and patient 3 were identified using next generation sequencing and confirmed with Sanger sequencing, whereas the variants in patient 2 and patient 4 were identified and confirmed by direct Sanger sequencing. A 3D structural modeling of the TNSALP constructed based on its sequence homology to the placental isozyme (PDB ID: 1EW2), was used to locate the missense mutation [12].
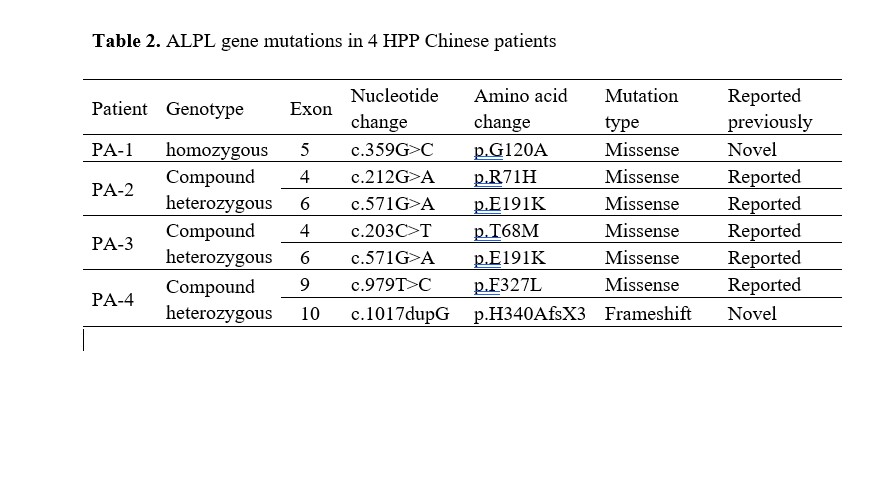
Six different variants were identified in the ALPL gene in our four patients, including five missense variants and one small insertion variant (Table 2). All variants were inherited from their unaffected parents except patient 1 (Figure 2). As the most severe and lethal infantile HPP in the present study, patient 1 carried a novel homozygous variant of c.359G>C (p.G120A) at exon 5 of the ALPL gene, which was predicted to be disease-causing by in-silico analysis of bioinformatics software. His father demonstrated a heterozygous status, while his mother was not involved in this study. To further investigate the effect of the novel missense mutation of c.359G>C, a 3D structure modeling of TNSALP was used to locate the residue, indicating that the residue p.G120A was located in the secondary structure of the TNSALP homodimer interface.
Patient 2, patient 3 and patient 4 were compound heterozygote at the ALPL gene. Patient 2, the childhood HPP, had been identified with two disease-causing missense variants, c.212G>A (p.R71H) at exon 4 and c.571G>A (p.E191K) at exon 6. As another childhood HPP, Patient 3 carried the identical c.571G>A (p.E191K) variant at exon 6 with Patient 2, and another pathogenic c.203C>T (p.T68M) variant at exon 4. Patient 4 was the mildest odonto HPP in the present study. He demonstrated pathogenic missense mutation c.979T>C (p.G120A), and a novel mutation c.1017dupG which was predicted to result in a translation frameshift and premature protein termination (p. H340AfsX3).
Literature review
In order to provide a comprehensive overview of Chinese patients with HPP, we reviewed all publications regarding Chinese HPP cases in the PubMed database (https://www.ncbi.nlm.nih.gov/pubmed) and the ALPL gene mutations database (http://www.sesep.uvsq.fr/03_hypo_mutations.php)[13-21].
The full spectrum of ALPL gene mutations in reported Chinese HPP patients is presented in Figure 3. A total of 26 mutations from 15 Chinese families were previously reported, including 20 missense mutations, 4 small deletion mutations, one small insertion and one splice site mutation. The 26 mutations were distributed throughout all 12 exons of the ALPL gene except for exon 1 and 8, with mutations most often located at exon 5 (27.6%, 8/29). Three mutations (c.407G>A, c. 1162T>C and c.1166C>A) were detected twice. No hot mutation was identified. The most common form of HPP in Chinese patients was childhood HPP, followed by adult HPP, whereas only one patient with onset of age at 1 month old was reported as infantile HPP. No lethal HPP has been reported in Chinese patients.
In the present study, three clinical forms of HPP were diagnosed in four Chinese children, which were infantile, childhood and odonto HPP. All six mutations (c.203C>T, c.212G>A, c.359G>C, c.571G>A, c.979T>C and c.1017dupG) were first reported in the Chinese population. Of which, c.359G>C and c.1017dupG were novel mutations. Patient 1 carrying homozygous c.359G>C exhibited severe phenotype of lethal infantile HPP.
{kind=link}
{kind=link}
{kind=link}