Cell line and cell culture.
B16 mouse melanoma cells obtained from iCell Bioscience Inc. (Shanghai, China) were cultured in RPMI 1640 medium supplemented with 10% fatal bovine serum (FBS) and 1% penicillin-streptomycin solution at 37 ℃ under 5% Carbon dioxide.
Mice.
4 to 6 weeks-old female C57BL/6 mice (14-18 g) were feed at the condition of 25 ℃ and 55% of humidity in Experimental Animal Center of Zhengzhou University. All animal studies were performed in accordance with the guidelines approved by Henan laboratory animal center. The license number of C57BL/6 is SCXK (xiang)2019-0004.
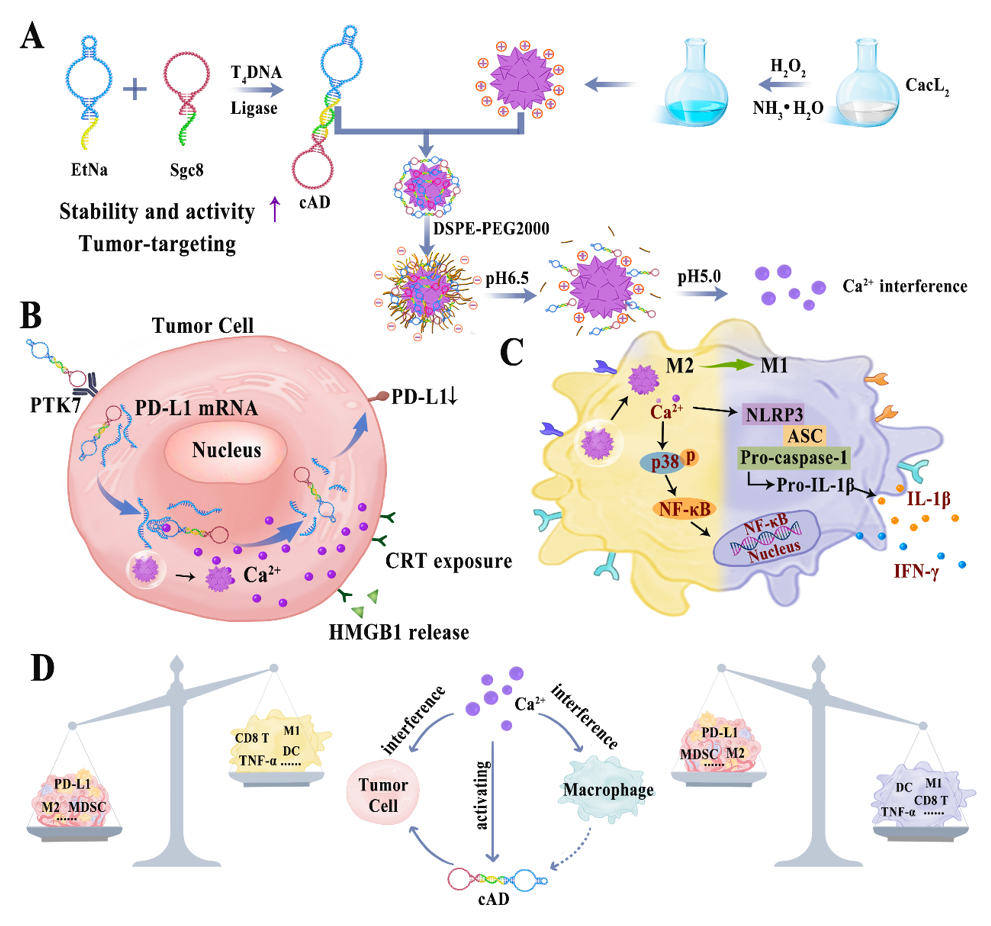
Synthesis of CaNP, cAD.
Synthesis of CaO2 nanoparticle (CaNP): Firstly, 3% hydrogen peroxide solution and 2 mol/L calcium chloride solution were prepared as the original solution. 1 mL of deionized water and 1 ml of calcium chloride were added to 60 ml of absolute methanol in turn. After stirring vigorously for 10 min, 1 ml of hydrogen peroxide was added. After stirring for 5 min, ammonia was added to the mixing system drop by drop until the color changed to Cambridge blue. Centrifugation at 8000 g for 20 min, repeated washing with methanol twice, and finally dispersed in deionized water.
Synthesis of circular aptamer-DNAzyme conjugate (cAD): 8 μL of EtNa (10 μM) and Sgc8 (10 μM) were added to 18 μL of DNase/Rnase-Free water, and then 10 × T4 DNA Ligase Buffer was added. After incubation at 95 °C for 5 min, the T4 DNA Ligase was rapidly cooled to 16 °C, and then added 2 μL of T4 DNA Ligase. After incubation at 16 °C for 12 hours, the above mixture was heated at 75 °C for another 5 min to obtian cAD.
Investigate the best feeding ratio of CaNP and cAD.
CaNP (5 mg/mL) and cAD (10 μM) were mixed by volume ratio of 1:1, 1:5,1:10,1:15, after incubating at room temperature for 4 hours, and 3% agarose gel was prepared to investigate the best feeding ratio of CaNP and cAD. CANP and cAD were used as the control.
Synthesis of CaNP@cAD-PEG.
CaNP@cAD-PEG was prepared by mixing cAD and CaNP in a volume ratio of 1:10. After incubation for 6 h at room temperature at 300 rpm/min, DSPE-PEG2000 was added in a mass ratio of 2:1 with CaNP. The mixture was shaken at room temperature for another 3 hours and then centrifuged at 8000 g for 20 min to obtain CaNP@cAD-PEG.
Characterization of Sgc8 aptamer targeting efficiency.
Sgc8 and random base sequences were labeled with FAM fluorescence, and 1µM of Sgc8 and random sequences were incubated with B16 cells at 37 ℃ for 2 h, respectively. Meanwhile, Sgc8 was co-incubated with HL-7702 cells as the control group, and the fluorescence intensity of each group was detected by a flow cytometer (BD Accuri®C6, American) and data analysis was performed with FlowJo (Version 10).
Cleavage ability of cAD in vitro.
cAD (100 nM) was incubated with an equal amount of PD-L1 mRNA (100 nM) at 37 ℃ for 12 h in Tris-HCl buffer (pH = 8.8) containing different concentrations of CaCl2 (1 mM, 10 mM, 20 mM, 30 mM, 40 mM and 50mM), respectively. After incubation, the cleavage efficiency was verified by polyacrylamide gel electrophoresis.
Study on the stability of cAD in vitro.
cAD (100 nM) was incubated with were added to 7.75 μL of DNase/Rnase-Free water, and then 10 × Exo I Buffer and 0.25 μL Exo I was added. After incubation at 37 °C for 1 h, 4 μL 6×DNA loading buffer was then added. The samples were then tested with 3% agarose gel. Meanwhile, Sgc8 and EtNa were also treated as control.
Characterization of CaNP, CaNP@cAD-PEG.
The morphology of CaNP and CaNP@cAD-PEG were detected via TEM (Tecnai G2 F20). The particle size potential is obtained by dynamic light scattering. 100 μg/mL of CaNP and CaNP@cAD-PEG nanoparticles in water solution were applied onto a 300-mesh carbon-copper grid. The excess solution was removed by filter paper. Images were recorded by a transmission electron microscope (JEM 1200EX, JEOL, Japan) operated at a voltage of 120 KV. 1 mL 50 μg/mL CaNP and CaNP@cAD-PEG in deionized water were used to measure the corresponding size distribution and zeta potential by a Zeta sizer (Nano ZS-90, Malvern, UK).
Characterization of CaNP@cAD-PEG in acidic environment
Morphology of CaNP@cAD-PEG in different pH values: CaNP@cAD-PEG (100 μg/mL) was fully dispersed into PBS buffer (pH 7.4, pH 6.5, pH 5.0) for 3 h, respectively. Then the above solution was dripped onto the copper network, and after removing the excess water, the morphology of nano particles was observed by a transmission electron microscope (JEM 1200EX, JEOL, Japan).
Ca2+ release from CaNP@cAD-PEG in acidic environment : CaNP@cAD-PEG (100 μg/mL) was fully dispersed into PBS buffer (pH 7.4, pH 6.5, pH 5.0) for 0.5 h, 1 h, 2 h, 4 h, 8 h, 12 h, respectively. Subsequently, the above mixtures were centrifuged at 12000 g for 20 min, the supernatant was collected for Ca2+ detection by inductively coupled plasma mass spectrometry (ICP-MS: Aglient 7800).
cAD release from CaNP@cAD-PEG in acidic environment: CaNP@cAD-PEG (cAD was labelled with FAM) was dispersed into different PBS buffer (pH 7.4, pH 6.5 and pH 5.0) at a final concentration of 1 mg/mL, and then incubation at 37 oC, 600 rpm under lucifuge environment for 0.5 h, 1 h, 2 h and 3 h, respectively. The fluorescence intensity was measured by a microplate reader (Ex: 488, Em: 525) (Synergy H1, BioTek, America). The release rate of cAD was calculated by ratio of fluorescence intensity to the total amount.
Cleavage ability of CaNP@cAD-PEG in vitro.
50 μg/mL of CaNP@cAD-PEG were incubated in PBS buffer (pH 7.4, pH 6.5 and pH 5.0) at 37 ℃ for 12 h, respectively. Subsequently, the above mixtures were centrifuged at 12000 g for 20 min, the supernatant was collected to incubate with PD-L1 mRNA (500 nM) at 37 ℃ for 12 h in Tris-HCl buffer (pH = 6.8), the cleavage efficiency was verified by polyacrylamide gel electrophoresis.
In vitro biodistribution of cAD in B16.
B16 in Confocal petri dish were incubated with cAD (labelled with FAM, 300 nM) for 2, 4 and 6 h. After incubation, cells were stained with Lyso-Tracker Red (75 nM) for 15 min. Then CLSM images were acquired after staining with Hoechst 33342 for 10 mins (TCS SP8, Leica, Germany).
Intracellular free Ca2+ detection in B16.
B16 in confocal petri dish were incubated with CaNP (35 μg/mL) for 0, 2 and 4 h, respectively. After incubation, cells were stained with Flou-3AM (1 μM) for 30 min. And then incubated with Hoechst 33342 for 10 min. Lastly, CLSM images were acquired for characterizing the intracellular free Ca2+.
Measurement of PD-L1 expression in B16.
qRT-PCR: cells were seeded at 6-well plates at 1×105 per well and cultured for 12 h, and then were incubated with different nanoparticles (CaNP, CaNP@cAD-PEG, CaNP@RcAD, cAD) for 12 h, 35 μg/mL nanoparticles or 300 nM cAD was applied, respectively. After washed with PBS, cells were incubated with fresh complete medium for another 12 h. And then, precipitate of cells was completely collected by trypsin digestion and centrifugation (2000 rpm, 5 min). Then mRNA was isolated with a Trizol Regent kit (Invitrogen). One microgramme of RNA sample was utilized to obtain cDNA. One microliter of cDNA sample and specific primers were used to amplify PD-L1 cDNA according to the manufacturer’s parameters (Custom gene qRT-CANPR Quantitation Kit) on a Real-Time CANPR machine.
Western blot analysis: cells were treated according to the above same method. The cells were collected and washed twice with PBS. Cell precipitations were lysed for 1 h at 4 oC. The protein content was measured by Bradford assay. Western blot analysis was conducted using standard method.
Generation of mouse BMDM.
The bone marrow cells were isolated from female C57BL/6 mice femurs and cultured with 20 ng/mL recombinant M-CSF for 6 days. On day 7, naive macrophages (BMDMs) were collected and then stimulated with 20 ng/mL IL-4 (MCE) plus 20ng/mL IL-13(MCE) or 100 ng/mL LPS (MCE) plus 20 ng/mL IFN-γ (MCE) for 24 h to generate the BMDM-M2 or BMDM-M1 macrophages, respectively.
Cytotoxicity assay of BMDM with different concentration of CaNP.
BMDM in 96-well plates was incubated in medium with different concentration of CaNP (1, 10, 20, 50, 100 µg/mL) for 24 h, respectively. And after washing with PBS, BMDM were incubated in medium with 10 µL Cell Counting Kit-8, after 3 h incubation, absorbance in 450 nm was detected. Cell activity (%) =[A(lactic acid)-A(blank)] / [A(lactic acid)-A(blank)]×100
Intracellular free Ca2+ detection in BMDM.
BMDM in confocal petri dish were incubated with CaNP (35 μg/ml) for 0, 2 and 4 h, respectively. After incubation, cells were stained with Flou-3AM (1 μM) for 30 min. And then incubated with Hoechst 33342 for 10 min. Lastly, CLSM images were acquired for characterizing the intracellular free Ca2+.
Characterization of polarization of macrophages induced by CaNP.
Western blot analysis: M2-BMDM in 6-well plates were treated with CaNP (35 μg/mL)/ H2O2 (0.7 μM)/ Cap (70 μg/mL) for 6 h, after washed with PBS, cells were incubated with fresh complete medium for another 42 h. At the same time, M2-BMDM pretreated with CsA (1 μM) for 2 h were treated according to the above method to confirm the effect of Ca2+on macrophages polarization. The cells were collected and washed twice with PBS. Cell precipitations were lysed for 1 h at 4 oC. The protein content was measured by Bradford assay. Western blot analysis was conducted using standard method.
Flow cytometry analysis: M2-BMDM in 6-well plates were treated with CaNP (35 μg/mL)/ H2O2 (0.7 μM)/ Cap (70 μg/mL) for 6 h, after washed with PBS, cells were incubated with fresh complete medium for another 42 h. The cells were collected and washed twice with PBS. Cells were incubated with CD206/CD86/CD11b/F4/80 antibody. And then fluorescence was detected by flow cytometry.
Cytokine secretion: M2-BMDM in 24-well plates were treated with CaNP (35 μg/mL)/ H2O2 (0.7 μM)/ Cap (70 μg/mL) for 6 h, after washed with PBS, cells were incubated with fresh complete medium for another 42 h, respectively. The cell supernatant of BMDM was collected into new 1.5 ml tubes, centrifuged at 300 g at 4 °C for 10 min to remove the sediment and detect immediately. ELISAs (EK2143/2-96 (Mouse CXCL9/MIG ELISA Kit), EK201B/3-96 (Mouse IL-1β ELISA Kit), EK212/3-96 (Mouse IL-12p70 ELISA Kit) and EK210/3-96 (Mouse IL-10 ELISA Kit) both from MULTISCIENCES (LIANKE) BIOTECH, CO., LTD) were performed according to standard protocols.
Immunofluorescence analysis of NF-κB nuclear transcription: M2-BMDM in 6-well plates were pre-treated with before treating with CaNP + CAT (CaNP: 35 μg/mL; CAT: 10 ug/mL) for 6h, cells were incubated with fresh complete medium for another 42 h. At the same time, M2-BMDM pretreated with were treated CsA (1 μM) for 2 h fluorescence to observe the effect of Ca2+ on NF-κB nuclear transcription.
Detection of macrophage phagocytosis: M2-BMDM in 6-well plates were treated with CaNP (35 μg/mL)/ H2O2 (0.7 μM)/ Cap (70 μg/mL) for 6 h, after washed with PBS, cells were incubated with fresh complete medium for another 42 h, respectively. Then, B16 cells were labelled with Cell Tracker Deep Red, BMDM were labelled with Cell Tracker Green. After incubation for 2 h, the cells were collected and washed with PBS for twice. And then fluorescence was detected by flow cytometry.
Detection the effect of macrophage polarization on T cell proliferation.
M2-BMDM in 6-well plates were treated with CaNP (35 μg/mL)/ H2O2 (0.7 μM)/ Cap (70 μg/mL) for 6 h, after washed with PBS, cells were incubated with fresh complete medium for another 42 h, respectively. Then, CD8+ T cell labelled with CFSE were co-incubated with cell supernatant of BMDM. After incubation for 48 h, CD8+ T cell were collected and washed with PBS for twice. And then fluorescence was detected by flow cytometry.
Activation of macrophage inflammasome.
M2-BMDM in 6-well plates were incubated with 500 ng/mL LPS for 4.5 h, after washed with PBS, M2-BMDM were treated with CaNP (35 μg/mL)/ H2O2 (0.7 μM)/ Cap (70 μg/mL)/ CaNP + CAT (CaNP: 35 μg/mL; CAT: 10 ug/mL) for 6 h, after washed with PBS, cells were incubated with fresh complete medium for another 42 h, respectively. The cell supernatants were collected to detect IL-1β according to ELISA procedure, and Cell precipitations were lysed for 1 h at 4 ℃. The protein content was measured by Bradford assay. Western blot analysis was conducted using standard method.
Biodistribution of CaNP in vivo.
Each BALB/c mouse was subcutaneously injected with B16 cells (1×107) on the left armpit. The tumor bearing mice were randomly grouped (n=3) when the tumor volume reached 200 mm3. The mice were then intravenously injected with 100 µL of CaNP (10 mg/mL) and saline as control . After 24 h of adimination, mice were then sacrificed for the major organs (heart, liver, spleen, lung and kidney) and tumors. After concentrated hydrochloric acid digestion for 12 h, the deionized water is diluted and filtered to collect the solution for Ca2+ detection by ICP-MS (Aglient 7800).
Biosafety evaluation of CaNP@cAD-PEG in vivo.
After treatment for 5 times, 5 mice from saline and CaNP@cAD-PEG groups were sacrificed by taking blood from the abdominal aorta and the obtained blood was used for blood chemistry analysis and blood routine analysis.
In vivo anti-tumor efficiency.
The B16 tumor-bearing mice were divided randomly into four groups: saline, cAD, CaNP, CaNP@cAD-PEG, aPD-L1, aPD-L1+CaNP (5 in each group). The different formulations were administrated through tail vein injection (cAD: 1 µM, 200 µL; CaNP: 5 mg/mL, 200 µL). The treatments were performed 7 times every 2 days. Body weights and tumor sizes were monitored every other day after the corresponding treatments. The tumor volume was calculated by the equation: Tumor volume = (width2×length)/2. Relative tumor volume was the corresponding changes relative to the initial value measured before treatment. All mice were photographed and euthanized at two weeks post injection, and the tumors, lymph nodes and spleens were taken out and photographed. The collected organs and tumors were fixed immediately in 10% paraformaldehyde solution, followed by standard dehydration and paraffin embedding. The embedded tissues were then sectioned into 4 μm slices and then subjected to standard H&E and Tunnel staining for histological analysis.
Dection of PD-L1 expression in B16 tumor.
The tumors were harvested 2 days post the final treatments and cut into small pieces to homogenate. After washing twice with PBS, Cell precipitations were lysed for 1 h at 4 oC. The protein content was measured by Bradford assay. Western blot analysis was conducted using standard method.
Detection of immune filtration in tumor.
The tumors were harvested 2 days post the final treatments and cut into small pieces randomly selected and weighed 0.04-1g (0.18 g), added 2.35 mL RPMI1640, 100 μL Enzyme D, 10 μL Enzyme R, 12.5 μL Enzyme A, and incubated at 37 °C for 40 min. Then filtered through a 70 μm filter, and 5 mL RPMI1640 was added to rinse the undigelled tissue.Wash it with PBS and suspend it again in 100 uL system. The cell suspension was stained with anti-CD11b-FITC, anti-CD80-PE, anti-CD206-PE for macrophage detection and anti-CD3-FITC, anti-CD45-PE/Cy7, anti-CD4-ACANP, anti-CD8α-PerCP, anti-FoxP3-PE for CD4/CD8/Treg detection. anti-CD3-FITC, anti-CD45-PE/Cy7, anti-CD8α-PerCP, anti-IFN-γ-APC for IFN-γ detection. anti-CD45-FITC, anti-CD11b-PE, anti-CD206-PerCp/cy5.5, anti-CD80-PE/cy7, anti-Gr-1-ACANP for M1/M2/MDSC detection. anti-CD45-PE/cy7, anti-CD11cACANP, anti-CD103-PE for DC detection. Flow cytometry acquisition was performed on a Cyan flow cytometer (BD LSR Fortessa) and data analysis was performed with FlowJo (Version 10).
Detection of the immune memory effect of CD8+T cells in spleen.
The mice were killed by dislocating executed 10 days post the distal tumor implantation, soaked in 75% ethanol for 10 min, and the limbs of the mice were fixed on the foam plate with the abdomen facing up with a needle. Pinch the abdominal skin with forceps, scissors cut a small mouth, expose the abdominal wall, tear the skinwith two forceps in the small mouth, try to expose the abdomen; The abdominal wall was clamped with forceps, the abdominal cavity was cut open, the spleen was removed, and the spleen was removed with an elbow forceps clamp. The removed spleen was placed in PBS and ground with the frosted surface of a sterilized and pre-wetted glass slide; The ground single cell suspension was filtered through a 70 μm cell screen to remove large tissue mass. The centrifuge was centrifuged at 300×g for 5 min in a cryogenic refrigerated centrifuge. Add erythrocyte lysate (3 times volume), gently beat evenly, and lyse for 5-10 min at room temperature; Wash with PBS 7.2 for 2 times, count, resuspend with PBS 7.2 (containing 1 mM EDTA and 2% FBS), adjust the cell density and stained with anti-CD3-FITC, anti-CD8α-PerCP, anti-CD44-APC and anti-CD62L-PE antibodies. Flow cytometry acquisition was performed on a Cyan flow cytometer (BD LSR Fortessa) and data analysis was performed with FlowJo (Version 10).
Analyze of the treatment-induced cytokine secretion in vivo.
The blood samples were agglutinated for 30 min at room temperature. Centrifuged at 1000 g for 10 min. Serum samples were collected for immediate testing. ELISAs (PT512 (Mouse TNF-α ELISA Kit) and P1580 (Mouse IFN-γ ELISA Kit), both from Beyotime) were performed according to standard protocols.
Immunofluorescence and immunohistochemical analysis of tumor tissues.
Frozen tumor sections were stained with immunofluorescence and immunohistochemical staining. The tumor was first placed in 4% paraformaldehyde (PFA) for 24 h at 4 ℃, and then transferred to 15% and 30% sucrose solution (w/w) for dehydration. The tumor was implanted in the optimal cutting medium (O.C.T.) and the frozen section was placed in a cryostat microtome. The dye was rinsed with PBS, permeated, and then sealed at room temperature with 5% bovine serum albumin (BSA) for 1 h, followed by staining with different major antibodies: CD4, CD8, CD206, CD80, DAPI overnight at 4 ℃, as per manufacturer's instructions. After adding fluorescently labeled secondary antibodies (goat anti-rat IgG and goat anti-rabbit IgG), the slides were analyzed by confocal microscopy.
Statistical analysis
Statistical analysis Statistics were performed using GraphPad Prism 8. Differences between two experimental groups were determined by two-tailed Student’s t test and multiple groups by one-way ANOVA with Tukey’s post-test. Survival curves were assessed with a log-rank (Mantel-Cox) test. TCGA gene data correlations were tested using nonparametric Spearman’s test. All bar graphs show means ± SEM. *P <0.05, **P < 0.01, ***P < 0.001.
Data availability
The authors declare that all the data supporting the findings of this study are available within the article and its Supplementary Information files, and from the corresponding author on reasonable request.
{kind=link}